Down syndrome, also known as trisomy
21, is the most common genetic disorder causing intellectual disability and the most
frequently occurring human chromosomal syndrome. Distinct features of the face,
hands, and feet are common. Other congenital anomalies (e.g., heart and
gastrointestinal defects) and acquired conditions (e.g., hypothyroidism, hearing
impairment, and celiac disease) occur more frequently in children with Down
syndrome.
Other Names & Coding
Down's syndrome
DS
Mosaic Down syndrome
Translocation Down syndrome
Trisomy 21
The prevalence of Down syndrome varies by age (due to the combined impact of
increasing life span and selective terminations decreasing birth prevalence) and
country (because of dramatic differences in availability of prenatal testing and
termination and population attitudes). The probability of having a baby with Down
syndrome increases as the mother’s age increases, but due to higher pregnancy rates
in younger women, most children with Down syndrome are born to women under 35 years
of age. Changes in childhood survival have impacted the age distribution of people
with Down syndrome, with more individuals living into their fourth, fifth, and sixth decades. The graph (left) is based on the
National Down Syndrome Cytogenetic Register and shows pooled prevalence (per 10,000
live births) of Down syndrome by maternal age. [Mai: 2013]
Prevalence would have been higher but for Down syndrome-related elective pregnancy
terminations. [de: 2017] Prevalence would have been higher
but for Down syndrome-related elective pregnancy terminations. A study in Atlanta
found that the frequency of elective terminations following an abnormal prenatal
cytogenetic test varied across race-ethnicity groups. Termination rates following
identification of trisomy 21 were 5.7% in Hispanic, 15.2% in non-Hispanic black, and
32.3% in non-Hispanic white populations. [Jackson: 2014]
The estimated birth prevalence of Down syndrome among live
births in the United States in 2010 was 1:826. The prevalence across all ages was
estimated to be 1:1,499, and the estimated prevalence adjusted for age distribution
in pediatric practice was 1:884. [de: 2017]
[Bocian: 1999] In 2014 in the United Kingdom, the overall
prevalence was 1:1574. [Alexander: 2016]
Genetics
Down syndrome refers to the phenotype or the pattern of physical traits, while
trisomy 21 represents the genotype, i.e., the underlying chromosomal basis of the
condition. In most individuals with trisomy 21, the additional chromosome results
from the sporadic occurrence of nondisjunction of chromosome 21 during meiosis
(>90% are of maternal origin). A small percentage (3-4%) of Down syndrome results
from an unbalanced translocation between chromosome 21 and another chromosome
(usually 13, 14, or 15). About 25% of these unbalanced translocations are familial;
the rest are sporadic. [Bull: 2022] A smaller percentage of
individuals with Down syndrome have trisomy 21 mosaicism from postzygotic
nondisjunction during mitosis of the fertilized egg or from postzygotic loss of a
chromosome 21 from a trisomic zygote. [Bull: 2022] In
mosaicism, only some of the cells in the body have the extra #21 (usually expressed
as a percentage of cells counted on the karyotype), and the remainder have the
typical 46 constitution.
Prognosis
The severity of co-occurring congenital anomalies and degree of associated
cognitive disability and social adaptability is variable. People with Down syndrome
have an increased risk for certain medical conditions such as congenital heart
defects, respiratory and hearing problems, Alzheimer’s disease, childhood leukemia,
and thyroid conditions. Scientific advances in health care for these conditions and
social advances in understanding the importance of educational and social
interventions have substantially improved the likelihood of a productive life for
individuals with Down syndrome. With the aid of a job coach, many adults with Down
syndrome are employed in the private sector. Life expectancy for people with Down
syndrome has increased dramatically in recent decades – from age 12 in 1949 to age
58 and older now. [de: 2017]
Bull MJ, Trotter T, Santoro SL, Christensen C, Grout RW, Burke LW, Berry SA, Geleske TA, Holm I, Hopkin RJ, Introne WJ, Lyons
MJ, Monteil DC, Scheuerle A, Stoler JM, Vergano SA, Chen E, Hamid R, Downs SM, Grout RW, Cunniff C, Parisi MA, Ralston SJ,
Scott JA, Shapira SK, Spire P. Health Supervision for Children and Adolescents With Down Syndrome. Pediatrics.
2022;149(5).
PubMed abstract / Full Text
Roles of the Medical Home
After the diagnosis of Down syndrome, the medical home should provide
acute-care treatment, well-child checks, and chronic-care visits. At chronic-care
visits, review progress, proactively manage problems, provide anticipatory guidance,
vaccinations, and other preventive services. The medical home is pivotal in
implementing screenings, evaluations, and interventions based on treatment
guidelines. Management focuses on maximizing the child's capabilities at home and
optimizing social inclusion. Treatment should start as early as possible, and the
medical home, in collaboration with the family, should initiate and coordinate
interdisciplinary care. Develop goals include optimizing growth and development and
providing ongoing information to families about available interventions, community
resources, evolving scientific understanding of trisomy 21, and emerging treatments.
The family should be central in all decision-making.
Clinical Assessment
Overview
Published guidelines for surveillance, screening, and caring for children with
Down syndrome tend to focus on high prevalence issues and areas where consensus can
be reached. [Bull: 2022] However, a number of issues are
not well-addressed, including gastroesophageal reflux disease, constipation,
frequent respiratory issues, and recognition of autism and ADHD. A comprehensive
review of systems and evaluation of identified issues are key at all visits.
Pearls & Alerts for Assessment
Sleep apnea is common
Sleep apnea occurs in up to 45% of individuals with Down syndrome. The
etiology may be obstructive, central, or mixed. A subset of individuals
exhibits clinically significant sleep apnea without overt signs of upper
airway obstruction. Children may have a normal sleep study and then have
significant apnea a few years later. When obstructive sleep apnea is treated
with a tonsillectomy and adenoidectomy, it improves but often does not
resolve the concerns. A repeat sleep study should be performed in any
situation where a child has new or persistent symptoms. See Sleep under
Comorbid Conditions below.
Atlanto-axial instability (AAI) may not require treatment
While 13-14% of patients with Down syndrome show evidence of atlantoaxial
instability (AAI) on X-ray, only 1-2% have symptoms that require treatment.
Treatment guidelines no longer recommend screening all patients with X-rays.
Rather, clinical care should focus on education for families regarding early
symptoms and monitoring for the emergence of clinical signs of AAI as
discussed under orthopedics. Careful questioning for symptoms and a
neurologic exam should be part of any sports physical exam. Community
organizations have yet to effect this change in guidelines, so some may
still require X-rays.
Wheezing may not be asthma
While wheezing is common, asthma is not usually an accurate diagnosis if
diagnostic criteria are accurately applied. [Watts: 2013] There should be careful consideration of other potential
causes.
Screening
For the Condition
Prenatal The 2016 American College of Obstetricians and Gynecologists
(ACOG) guidelines advise offering screening, diagnostic testing options, and
counseling about the risks for aneuploidy during every pregnancy prior to 20
weeks of gestation. Testing should result from informed patient choice; women
have the right to decline genetic screening in a shared-decision making model.
Maternal age and other risk factors must guide the interpretation of any test
results. [Committee: 2016]
[Bull: 2022]
First trimester screening, available in weeks 10-13 of
gestation, includes nuchal translucency (NT) measured by ultrasound,
testing maternal blood for levels of pregnancy-associated plasma protein
A levels (PAPP-A) and serum-free beta or total human chorionic
gonadotropin (hCG).
NT measurement can help assess individual
fetuses in multi-fetal pregnancies (e.g., twins) but is not
considered sensitive or reliable as a single
test.
The Triple screen, available from 15-22 weeks of
gestation, includes maternal blood levels of hCG, unconjugated estriol,
and α-fetoprotein (AFP). Used in isolation, this is the least accurate
screening method.
The Quad screen, available from 15-22 weeks of
gestation (ideally in weeks 16-18), incorporates assessing maternal
blood levels of hCG, unconjugated estriol, α-fetoprotein (AFP), and
dimeric inhibin A levels. The "Penta" screen adds measurement of levels
of hyperglycosylated hCG (also known as invasive trophoblast
antigen).
Integrated screening is a 2-step process that combines
NT measurement with serum measures from the triple and quad screening.
"Serum integrated" utilizes the same blood screening but does not
include NT. Results of integrated screening are not available until the
second trimester.
Sequential (stepwise or contingent) screening is like
integrated screening, except the first trimester risk assessment is used
to tailor subsequent screens and diagnostic testing. This approach
enables decision-making in the first trimester.
Cell-free DNA testing has rapidly become the most used screen for
aneuploidy. It is performed starting at 10 weeks and is the only screen
available for use in the third trimester. This method analyzes segments
of placental DNA found in maternal blood. It can be quite effective at
detecting trisomy 21 in high-risk populations (those with high pre-test
probability), but false positives are increased in low-risk populations.
In the last 5 years in the US, noninvasive prenatal screening (NIPS)
using cell-free DNA testing has replaced most of the modalities
mentioned above. Sensitivity, specificity, and positive predictive
values have increased in recent analyses. [Badeau: 2017]
Pre-implantation genetic screening can be used during
in vitro fertilization (IVF).
None of these screening tests are considered diagnostic. Mothers with
positive screens should be offered diagnostic testing with amniocentesis. Diagnostic evaluations are more invasive because they
require obtaining fetal tissue samples for genetic testing. Rates of loss
associated with these procedures are 0.1% to 0.3% when performed by experienced
providers. [American: 2016] Chorionic villus sampling
is done at 10-13 weeks, or amniocentesis is usually performed at 15-20 weeks but
can be done later. Testing embryonic tissue is also feasible prior to
implantation during in vitro fertilization.
Of Family Members
In standard trisomy 21, the risk of recurrence for parents of a child with
Down syndrome where the mother is less than 35 years of age is 1-2%. The reason
for this increased risk is unknown; the figure is empirically determined. If the
mother of the child with Down syndrome is >35 years, then the risk of recurrence
is considered the same as the specific maternal age risk for the population.
Less than 5% of confirmed Down syndrome diagnoses are due to a translocation and
potentially carry a higher risk of recurrence. [Bull: 2022] If a child with Down syndrome has a chromosomal
translocation, it is important to determine if the parents carry a balanced
translocation in order to provide counseling about the recurrence risk in future
pregnancies and determine if other family members should be
tested.
For Complications
The most recent guidelines [Bull: 2022] provide
recommendations for universal and symptom-based screening of newborns and
children with Down syndrome for a wide range of comorbid conditions. These are
compiled in the Down Syndrome Checklist (2022) ( 487 KB) for
use in practice. Newborn period Universal screening
Routine newborn screening
Hearing screening is adequate if ABR or OAE is
normal; if abnormal, the child should be referred for follow-up
evaluation.
Thyroid screening with thyroid stimulating
hormone (TSH) is adequate; if the routine screen uses only T4, a
TSH should be obtained during the nursery stay.
Echocardiogram to evaluate for congenital heart disease
(a normal fetal echo is not adequate since some heart defects can be
missed, in the stable infant, the echo can be completed electively in
the first weeks of life).
Hematologic screening: Complete blood count to assess
for a leukemoid reaction, myeloproliferative disorder, and polycythemia.
Vision screening: Evaluate for cataracts with the red
reflex exam.
Symptom-based screening
Consider a car seat trial in infants with low birth
weight, heart disease, or severe hypotonia.
Consider a modified barium swallow study for newborns
with feeding difficulties, respiratory concerns, or severe
hypotonia.
Assess for anorectal atresia/stenosis or Hirschsprung
disease in newborns who fail to pass meconium within the first 48 hours
of life.
If clinical concerns suggest their potential, assess
for intestinal atresia, airway abnormalities, obstructive sleep apnea,
and gastroesophageal reflux disease.
Routine screening for renal and urinary tract
abnormalities, atlantoaxial instability, and spinal anomalies is not
recommended unless indicated by symptoms (e.g., a urinary tract
infection, symptoms of urinary obstruction, persistent head tilt, or
torticollis).
Infancy through adolescence (refer to Down Syndrome Checklist (2022) ( 487 KB) for details and frequency of
recommended screening by age). Special considerations for children with Down
syndrome include:
Hearing loss from middle ear effusion, which is often
hard to visualize, is common. Some clinicians refer to ENT for routine
exams to aid in monitoring.
Eye examinations should include assessment of red
reflex. Due to the high prevalence of problems (e.g., myopia, hyperopia,
cataracts, strabismus, nystagmus), all children with Down syndrome
should be examined by a pediatric ophthalmologist by 6 months of age.
In-office photo screening is recommended, if available, after one year
of age.
All children with Down syndrome should be referred to
an Early Intervention program. Bright Futures guidelines recommend
standardized developmental screening (e.g., ASQ or PEDS) at 9 months of
age. This should be done for any child with Down syndrome who has not
been previously identified as having delays. [Committee: 2017] As many as 20-30% of children with Down syndrome
will have autism spectrum disorder; clinicians should monitor for
suggestive symptoms and refer for evaluation when indicated.
Radiologic imaging for atlantoaxial instability (AAI)
or spinal anomalies should be performed if symptoms suggest (e.g.,
persistent head tilt or torticollis, lower extremity increased
reflexes). All children should be managed with the potential for AAI
when positioning for intubation. Parents should be taught recommended
activity restrictions (e.g., do not teach tumbling, no head-first
diving, no trampoline till age 6 years and then only with supervision)
and clinical signs that suggest a need for evaluation (e.g., increased
tone in legs, change in gait or hand function, chronic headaches or neck
pain, chronic head tilt).
A sleep study is recommended by age 4 but should be
completed sooner if any symptoms suggest obstructive or central sleep
apnea.
Consider testing for celiac disease if there is failure
to thrive, chronic diarrhea, persistent constipation, chronic bloating,
or iron deficiency; it is unlikely if the child has not started foods
with gluten and is uncommon before age one.
Renal/urologic studies if concerns for obstruction
(e.g., posterior urethral valves) or urinary tract infection.
Presentations
Current prenatal screening will identify approximately 85-90% of fetuses with
Down syndrome. A European study found that roughly 90% of mothers of affected
fetuses opted to terminate pregnancy. [Morris: 2009] A
meta-analysis of US studies from 1995-2011 found a weighted mean termination rate of
67% among 7 population-based studies. [Natoli: 2012] The
birth of infants with Down syndrome not previously identified is becoming less
common but will continue. Clinicians may be considering Down syndrome when a newborn
is noted to have atypical features, hypotonia, or a major malformation associated
with Down syndrome. An infant's presentation may be subtle, occasionally leading to
missed diagnoses in the newborn period. These infants are likely to be recognized by
the primary care clinician due to poor growth, feeding concerns, developmental
delays, hypotonia, or concern for a medical condition associated with Down syndrome.
Guidelines for communicating a diagnosis of Down syndrome in
the prenatal and postnatal settings can assist the provider when preparing for an
informing interview. See [Skotko: 2009];
[Skotko: 2009]. Ten percent of individuals with Down syndrome
are identified after 1 week of age, and more than half of those not until adulthood.
Identification rates reflect past screening approaches; it is unclear how updated
screening guidelines will impact this. Delayed diagnosis is more likely when an
individual has mosaic trisomy 21, in which physical features may be subtle. Mosaic
trisomy 21 has been identified in adult individuals with intellectual challenges but
no physical features of Down syndrome.
Diagnostic Criteria
While physical features may suggest a diagnosis of Down syndrome,
confirmation requires chromosome analysis, which, in most individuals, will reveal
an extra chromosome 21. In 4% of patients with Down syndrome, analysis will find the
attachment of an extra-long arm of chromosome 21 to another chromosome
(translocation Down syndrome). A small percentage have an extra chromosome 21 in
only some of their somatic cells due to loss of the extra chromosome in mitosis or
nondisjunction during mitosis of the fertilized egg (mosaic Down
syndrome).
Differential Diagnosis
Other genetic syndromes with overlapping features include
Smith-Magenis syndrome, Zellweger syndrome, multiple X syndromes (e.g., 49,XXXXY),
and Noonan syndrome. Since many physical features of Down
syndrome occasionally occur in typical infants, the clinician should look for a
constellation of findings when considering the diagnosis. Epicanthal folds,
protruding tongue, single palmar crease, widely-spaced first and second toes,
hypotonia, and upslanting palpebral fissures are suggestive, especially in
combination; however, these features can all be found in people with normal
chromosomes. These issues underscore the importance of genetic testing to confirm
diagnosis.
Comorbid & Secondary Conditions
Individuals with Down syndrome are at risk for many associated
conditions, discussed below by organ system. Cardiovascular Congenital heart defects are found in around 50% of
infants with Down syndrome. [Watts: 2013]
[Bull: 2022] Early mortality is associated with the
presence of a cardiac defect, particularly if combined with a gastrointestinal
malformation. The most common defects include:
Atrioventricular septal defects, with or without other
heart defects (45%)
Ventricular septal defects, with or without other heart
defects (35%)
Isolated secundum atrial septal defect (8%)
Isolated persistent patent ductus arteriosus (7%)
Isolated tetralogy of Fallot (4%)
Other (1%)
Pulmonary hypertension may be diagnosed at birth or develop
in the child with unrecognized or untreated sleep apnea or heart defect. If
untreated, over the long term, pulmonary hypertension may not be reversible in the
patient with an unrepaired heart defect; Eisenmenger syndrome may evolve (in which
pulmonary hypertension, reversal of flow, and cyanosis develop as the left-to-right
shunt switches to right-to-left (due to rising pulmonary pressures). Acquired valvular dysfunction is common in adults with Down
syndrome without congenital heart disease (up to 50%).
Mitral valve prolapse is most common.
Tricuspid, aortic, and mitral insufficiency have also been
reported.
Nutrition
Newborns with Down syndrome are at risk for feeding
problems due to a weak suck, low tone, and problems related to organ
malformations. However, many mothers who breastfeed are successful if they
continue to try, especially with the guidance of a breastfeeding coach.
[Aumonier: 1983]
[Bull: 2022] Some infants will need significant
support during the first few weeks of life to attain success with breast or
bottle.
Older infants may have lingering tongue thrust, which can
delay success with the introduction of solids.
Oral aversions are common.
Self-feeding skills are often delayed due to delayed fine
and oral motor skills, oral aversions, and behavioral challenges.
Older children are at risk for excessive weight gain, which
may be more due to family health behaviors than a characteristic of Down
syndrome. [Bertapelli: 2016]
Behavioral feeding concerns, celiac disease, reflux,
chronic constipation, and diabetes are relatively common and result in an
additional need to focus on nutrition.
Respiratory Children with Down syndrome are at increased risk for recurrent acute
respiratory illness, including pneumonia, aspiration, bronchiolitis syndromes,
croup, and/or chronic lung disease. They are also at increased risk for
sleep-related breathing disorders, including obstructive and central apnea.
Contributing factors may include:
Respiratory infections, such as pneumonia and
bronchiolitis, are second only to congenital heart disease as causes for
hospitalizations and are associated with increased morbidity and mortality
compared to other children hospitalized for the same infections.
Structural abnormalities - midface hypoplasia, a normal
sized tongue filling a small oral cavity (thus looks large), small
subglottic area, laryngomalacia, narrow nasopharynx or choanal atresia,
enlarged tonsils and adenoids, tracheobronchomalacia, esophageal atresia,
and tracheal stenosis. In the lungs, abnormal pulmonary vasculature, reduced
number of alveoli, and enlarged alveolar ducts and alveoli may predispose
these children to increased respiratory problems. Subpleural lung cysts are
more commonly present, although the clinical relevance is unclear.
Congenital heart disease can also predispose children to respiratory
problems due to the complex interdependence of these body systems.
[Watts: 2013]
Immune deficiencies – both cellular and humoral immune
differences have been described. Immunoglobin G subclasses 2 and 4 have been
deficient in some children who have a normal total IgG level.
Gastroesophageal reflux or dysphagia leading to aspiration
Wheezing may be common, but asthma is not a likely diagnosis.
[Watts: 2013] It is postulated that the wheezing stems
from congenital lung abnormalities, tracheomalacia, upper airway collapse secondary
to hypotonia, or congenital heart disease. [Watts: 2013]
High-altitude pulmonary edema (HAPE) can occur in children
with Down syndrome, especially, but not limited to those with underlying congenital
heart defects, left-to-right shunts, and/or pulmonary hypertension. HAPE can be an
initial sign of pulmonary hypertension. [Watts: 2013]
Sleep Sleep apnea occurs in up to 45% of children with Down
syndrome, most commonly obstructive (50-80%), although central apnea can occur.
[Bull: 2022] Contributing factors may include
structural abnormalities (as mentioned under Respiratory above), tonsillar/adenoidal
hyperplasia, hypotonia, obesity, and brainstem dysfunction. Symptoms can include:
Abnormal breathing patterns in sleep
Snoring
Abnormal sleeping positions (e.g., sitting up)
Fragmented sleep (sometimes without snoring)
Inattention
Daytime sleepiness
Difficult morning arousal (due to carbon dioxide retention)
Early morning headaches (due to carbon dioxide retention)
Nocturnal enuresis
Failure to thrive
Behavioral problems
Behavioral sleep problems, including difficulty going to
sleep and staying asleep, also occur in many children with Down syndrome (as well as
in kids with normal chromosomes). Providing support to parents to help them optimize
sleep hygiene and manage behavioral sleep issues can be very helpful. Gastrointestinal The incidence of gastrointestinal anomalies in Down syndrome is
higher than in the non-DS population. The most common are:
Intestinal atresia (12% of individuals with DS)
[Bull: 2022]
Celiac disease. (~5%) Note that about a third of
individuals with Down syndrome and celiac disease have no overt clinical
symptoms. [Bull: 2022]
[Pavlovic: 2017] See Celiac Disease for more detail.
Hematologic Transient asymptomatic blood count abnormalities, including
neutrophilia, thrombocytopenia, and polycythemia, can occur in neonates with trisomy
21. Ten percent of infants with Down syndrome develop transient myeloproliferative
disease (characterized by the presence of blasts on the smear) with spontaneous
regression in the vast majority. [Bull: 2022] However,
transient myeloproliferative disease can cause significant morbidity/mortality due
to rare liver/heart failure, sepsis, DIC, and hyperviscosity, and 20-30% of children
with Down syndrome and myeloproliferation develop lymphoid or myeloid leukemia
(often delayed with a mean age onset of 20 months). [Dixon: 2006]
Individuals with Down syndrome are at increased risk for
leukemia throughout childhood. Despite the increased
incidence, the development of leukemia is still a relatively rare event (~1% of
individuals with Down syndrome), and routine screening (beyond a complete blood
count with differential at birth) is not recommended. However, follow-up CBCs over
the subsequent 36 months is important for any child who has a leukemoid reaction or
evidence of myeloproliferative disease on their newborn CBC, even if it appears to
normalize in the newborn period. Screening for
iron deficiency is recommended in children with trisomy
21 yearly. Hemoglobin and RBC indices perform poorly (insensitive) due to baseline
mild macrocytosis in many children with Down syndrome. Guidelines suggest checking a
taking a dietary/medical history and checking a hemoglobin in all patients. If the
dietary/medical history identifies any risk for iron deficiency, a marker of iron
storage should be measured (e.g., reticulocyte hemoglobin, ferrite, serum
transferrin). [Dixon: 2010]
Neurology
Microcephaly is relatively common in trisomy 21.
Seizures prevalence in children with Down syndrome is
1-14%. [Barca: 2014] Seizure types may include
infantile spasms (West syndrome), generalized tonic-clonic seizures, partial
seizures, Lennox-Gastaut, and reflex seizures. Of note, some children with
Down syndrome and no clinical seizures have EEG abnormalities that may
complicate interpretation.
New-onset of focal weakness is relatively common and has a
broad differential diagnosis. Etiologies found in a review of ten cases
included: infarction related to Moyamoya, vaso-occlusive disease, or venous
sinus thrombosis, traumatic subdural hematoma, brain abscess, spinal cord
injury (from cervical spine stenosis and/or atlanto-axial instability, and
brachial plexus injury. [Worley: 2004]
Dementia - By age 40, Alzheimer-type neuropathologic
abnormalities are found in all patients with Down syndrome, with or without
clinical dementia. [Lott: 2012] More than half of
individuals older than 50 years develop Alzheimer disease. Early-onset
epilepsy is associated with decreased risk of Alzheimer, whereas late-onset
epilepsy is associated with increased risk. [Menéndez: 2005]
Myelopathy may result from atlanto-axial instability or
subluxation of the occiput on C1 due to the ligamentous laxity seen in some
patients with trisomy 21.
Mental Health/Behavior “Dual diagnosis” refers to the co-existence of intellectual
disability and a psychiatric disorder, which affects 18-38% of individuals with Down
syndrome. [Capone: 2006] Comorbid neuropsychiatric
disorders include ADHD, autism spectrum disorder, stereotypical movements,
oppositional defiant and disruptive behavior disorders, anxiety, depression,
obsessive-compulsive disorder, and, rarely, psychosis. A summary of behavioral
disorders, their presentation and treatment can be found in the following articles:
[Dykens: 2007]
[Capone: 2006] Other sections of the Medical Home Portal
may be helpful, including those on Autism Spectrum Disorder, Anxiety Disorders, Depression, and Attention-Deficit/Hyperactivity Disorder (ADHD). Concerns about focus, attention span, activity level, and/or
impulsiveness (Attention deficit disorder) are common. In a 2017
prevalence study in Sweden, about 1/3 of children with Down syndrome met diagnostic
criteria for ADHD. [Oxelgren: 2017] Of these, many also met
criteria for an autism spectrum disorder. The following should be considered in the
evaluation of attention problems:
Hearing deficits
Vision deficits
Thyroid disorders
Sleep problems (e.g., sleep apnea can contribute to
impaired attention)
Impaired expressive communication
Education setting not appropriate for cognitive level or
learning style
Emotional problems (e.g., depression, anxiety)
Auditory processing disorders
These issues are summarized in Attention Problems in Down Syndrome: Is this ADHD? (1998, yet still
useful) by Dianne McBrien, MD, a developmental pediatrician at Children's Hospital
of Iowa. Autism spectrum disorder (ASD) in children with Down syndrome
is more common than previously thought when using earlier diagnostic criteria. It
may be as high as 40% in children with Down syndrome and is also more common in boys
than girls. [Oxelgren: 2017] Many children with Down
syndrome and autism also met criteria for ADHD. Standardized autism rating scales
have not been validated in those with Down syndrome. It is therefore recommended
that existing DSM criteria be rigorously applied over multiple observations in
different settings. Regressive autism has been noted to occur in children with Down
syndrome at an older age than seen in children without Down syndrome who have an
autistic regression. The accuracy or utility of a comorbid diagnosis of autism
spectrum disorder in children with profound cognitive impairment (IQ<25) has been
questioned. Although confirming an autism diagnosis may be more challenging in the
setting of Down syndrome, a separate autism diagnosis is likely to help in accessing
interventions such as applied behavioral analysis. Depression rates in individuals with Down syndrome range from
0-11% of individuals. This rate is about the same as in the general population and
in contrast to previous beliefs that people with Down syndrome are at higher risk
for depression. Onset of depressive symptoms tends to occur in adulthood, although
they can occur earlier and should be distinguished from hypothyroidism, hearing
impairment, and dementia. [Walker: 2011]
Ears/Hearing Hearing loss may be sensorineural, conductive, or mixed in
etiology and occur at higher rates in children with Down syndrome. Children 3-5
years old with Down syndrome have a 50-70% risk of chronic serous otitis media.
[Bull: 2022] During early development, even minor
hearing impairment can negatively impact language and cognitive development - by
adulthood, 60-80% have hearing loss. Sino-pulmonary disease, including recurrent/chronic sinusitis and
otitis, occur at higher rates. Children with Down syndrome may also
have auditory processing deficits that affect word perception, short-term auditory
memory, and sequential auditory memory. The following may be helpful for families:
Hearing and Vision Loss Associated with Down Syndrome (TSBVI). Eyes/Vision Individuals with Down syndrome are at risk for: [Bull: 2022]
Nystagmus
Strabismus
Cataracts, including congenital and
acquired
Lacrimal duct obstruction
Refractive errors (50% of children ages 3-5) leading to
amblyopia
Keratoconus (cone-shaped cornea)
All children with Down syndrome should be followed by an
ophthalmologist starting at 6 months of age. Dental Children and young adults with Down syndrome are at risk for:
[Bull: 2022]
Significant delay in eruption of both primary and secondary
teeth
Missing and/or malformed teeth
(hypodontia)
Dental crowding and overbite
Periodontal disease, developing in teen years may be
rapidly progressive [Bagić: 2003]
Halitosis
Cheilosis from chronic oral breathing
Aphthous ulcers
Oral candidal infections
Necrotizing ulcerative gingivitis
Providing preventive dental care may be difficult because:
Cognitive and fine motor skills may limit the child's
ability to perform brushing and flossing.
Anatomy (small mouth) and oral aversions may make it
difficult for others to provide care.
Behavioral and health issues (e.g., sleep apnea, congenital
heart disease) may increase the risk of sedation in the dental setting
.
Abnormalities in the roots of the teeth may impact
orthodontic planning.
A helpful handout for parents: Dental Care for the Patient with Down Syndrome. Endocrine Thyroid disorders, both congenital and autoimmune
hypothyroidism, occur with increased frequency. Congenital hypothyroidism occurs in
about 1% of children with Down syndrome. [Bull: 2022] Many
children develop euthyroid autoimmune disease, evidenced by a slightly elevated TSH,
normal free T4 level, and positive thyroid antibodies. Although opinions vary about
if and when to treat this, most endocrinologists do not treat if the child is
growing well and the TSH is less than 10-12. These children should be monitored, but
it may be many years before they develop a need for medications. Younger children
require more frequent monitoring given the impact of hypothyroidism on brain development. Short stature is common. The current guidelines recommend
using standard growth charts rather than Down syndrome charts; however, subsequently
updated growth charts for children in the US were published that have improved
characteristics. [Zemel: 2015] These updated height,
weight, and OFC growth charts are quite useful. Autoimmune Disorders The following are associated with Down syndrome:
Celiac disease (~5%): [Bull: 2022]
[Book: 2001] Note that about a third of individuals
with Down syndrome and celiac disease have no overt clinical symptoms.
[Pavlovic: 2017] See Celiac Disease for more
detail.
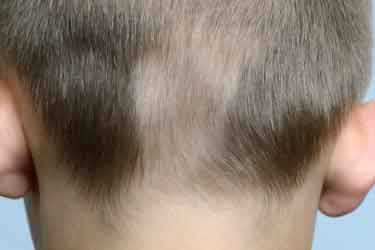
Alopecia areata (photo, right):
Asymptomatic non-scarring hair loss with spontaneous remissions and
exacerbations, often in combination with vitiligo. Alopecia may be localized
or may involve the entire scalp or body. Children with alopecia/vitiligo
should be carefully evaluated (history and physical) to identify any other
associated autoimmune conditions.
Many other disorders thought to be autoimmune in nature have
been reported, including multiple sclerosis, demyelinating neuropathy, and systemic
lupus erythematosus. Though more common in adults, these disorders have been
reported in children. The mechanisms for autoimmune disease in Down syndrome are
poorly understood. They may occur in combination and are more common in patients
with certain HLA markers. In childhood, screening is only recommended for thyroid
dysfunction and those with symptoms suggestive of celiac disease. However, the
clinician should monitor for signs and symptoms of all of these conditions and
assess when indicated. Orthopedics/Skeletal Hypotonia, ligament laxity, and increased joint flexibility lead to
orthopedic concerns. Individuals may also exhibit skeletal differences, such as a
thin, weak acetabular capsule, femoral anteversion, and a deficient posterior
superior acetabulum that may contribute to orthopedic problems. Orthopedic issues include:
Hip: Up to 8% may have hip problems, including
developmental dysplasia of the hip (in this population, hip problems may
begin after skeletal maturity and may significantly affect functional
ambulation), avascular necrosis, and slipped capital femoral epiphysis.
Lower leg: Genu valgum, patellar dislocation
Feet: Planovalgus, metatarsus primus varus, hallux valgus
Increased risk for low bone density and vitamin D
deficiency
Brachycephaly: though this does not cause functional
problems, families may request evaluation by craniofacial specialists to
consider molding orthotics
Atlantoaxial instability: While 13-14% of patients with
Down syndrome show evidence of atlantoaxial instability on X-ray, only 1-2%
have symptoms that require treatment. Parents should be educated to notify
their physician if their child has:
Neck pain
Persistent head tilt
Intermittent or progressive weakness
Changes in gait or loss of motor skills
Loss of bowel or bladder control
Increased or further decreased muscle tone in the
legs
Changes in sensation in the hands or feet
Dermatologic Individuals with Down syndrome are at risk for:
Atopic dermatitis
Syringomas
Benign skin tumors arising from sweat glands commonly about
the eyes/face
Norwegian scabies (crusted scabies)
Xerosis
Milia-like idiopathic calcinosis cutis
Skin infections, such as bacterial or fungal folliculitis
Elastosis perforans serpiginosa: deep red raised lesions
often occurring about the neck, chest, and arms
Angular cheilosis
Vitiligo
A number of benign dermatologic differences are also described, including:
Acrocyanosis in the newborn
Cutis marmorata (may be present up to several months of age
in infants with Down syndrome)
Hyperkeratosis of palms and soles
Urologic The following conditions have been reported in infants with Down syndrome:
Renal hypoplasia
Hydro-uretero-nephrosis
Uretero-vesical and uretero-pelvic junction obstruction
Vesico-ureteral reflux
Posterior urethral valves
Cryptorchidism
Testicular cancer
Infertility (present in most but not all males with Down
syndrome)
Developmental Children with Down syndrome are at increased risk for:
Delayed gross and fine motor development: Motor delays are
secondary to hypotonia, ligamentous laxity, decreased muscle strength, and
altered body proportions (shorter arms and legs).
Social-emotional developmental concerns: Delays in
social-emotional development can occur especially in children with Down
syndrome who also have an autism spectrum disorder
Sensory integration concerns: Many children are sensitive
to touch, manipulation, and textures in and around their mouth and other
parts of their body. Sensory issues may affect oral motor skills, the
ability of the parents to care for the child (e.g., dental and facial
hygiene, feeding), and the willingness/ability of the child to eat a variety
of tastes and textures. Sensory integration issues may be important in
expressive communication since producing communicative responses requires
processing and integrating sensory input.
Oral motor impairments: In early development, infants may
be at risk for inadequate nutrition due to poor oral motor coordination and
low tone. As children mature, additional oral motor problems may include
difficulty with different textures, drooling, and verbal apraxia.
Cognitive concerns: Intellectual disability impacts the
majority but not all individuals with Down syndrome. The degree of
intellectual disability can vary from mild to profound. Additional cognitive
processing concerns are common although often hard to define (e.g., slow
processing, working memory deficits, challenges with understanding
sequences).
Executive function problems: Can present as symptoms
consistent with ADHD, a common comorbid condition
History & Examination
The initial evaluation of the child with suspected Down syndrome should focus
on the prenatal, medical, and developmental history, as well a complete physical
and developmental evaluation as outlined below.
Follow-up visits should begin with open-ended questions
about patient/family concerns and issues. Review progress since last seen and
intercurrent illness or evaluations, specific symptoms, and current treatment
plan for underlying conditions (e.g., cardiac, thyroid, gastrointestinal) should
be reviewed.
Current & Past Medical History
Document past and present comorbid conditions, including
prior evaluative (e.g., cardiac echo, swallow study, sleep study) and surgical
procedures. A full review of symptoms is helpful, given the myriad comorbid
conditions associated with Down syndrome. Of particular concern are
cardiorespiratory, sleep, feeding, gastrointestinal symptoms, learning/behavior,
signs or symptoms of myelopathy, and concerns regarding hearing and
vision.
Family History
A 3-generation pedigree is indicated, though a family history of Down
syndrome or another chromosome abnormality is unlikely. The incidence of
aneuploidy in offspring increases with parental age, particularly maternal age.
A family history of pregnancy loss, especially miscarriages, can suggest a
familial translocation.
Pregnancy/Perinatal History
The pregnancy and perinatal history may include:
Abnormal prenatal ultrasounds (e.g., polyhydramnios, suggesting duodenal
obstruction, or minor ultrasound findings, such as redundant nuchal skin
and increased nuchal translucency)
Abnormal first- and second-trimester maternal screening (including
confirmation of diagnosis by amniocentesis or chorionic villus sampling
in some patients)
Detection of structural defects (including the prenatal diagnosis by
ultrasound of cardiovascular malformations or duodenal
atresia)
Developmental & Educational Progress
The child's functional abilities are key to
management. Assess the child’s method and level of expressive communication and
his/her understanding of language. Many children with Down syndrome have
significantly higher receptive than expressive language abilities, which is
sometimes related to verbal apraxia or ASD. Typical Down syndrome language
milestones include:
Smiling by 2 months (SD 1.5-4 months)
Verbalizing single words by 16 months (SD 9-31 months)
Verbalizing early phrases by 28 months (SD 19-96
months)
Average ages for attainment of gross motor skills in Down
syndrome include:
Rolling stomach to back by 6 months
Rolling back to stomach by 7 months
Sitting independently at 11 months
Belly crawl (>5 ft) by 14 months
Pull to stand from hands and feet by 17 months
Independent standing (>10 sec) by 21 months
Walking (15-20 ft) by 26 months
There is wide variation around these averages, and the child
with ongoing medical issues (e.g., repeated illness or surgeries) may exhibit
further delays. However, it is a pitfall to blame excessive delays on medical
issues without carefully considering the potential for comorbid conditions such
as hearing loss or autism. Review the services the child receives through early
intervention, the school district, or private therapy providers, the child's
rate of progress, and parents' satisfaction with current services.
Inquire about family, teacher, therapist, and other
caregiver concerns regarding development, attainment of functional goals, ADHD
or ASD symptoms, and/or behavioral challenges. Skills in activities of daily
living, eating, and community integration should be discussed with goals set for
each. Behavior challenges are common, including sleep
and feeding concerns, internalizing/externalizing behaviors, and poor social
inclusion. Consider whether problem behaviors and their frequency and intensity
are consistent with the child's functional abilities. The 5-year-old whose
receptive language skills are at the 3-year level is likely to have temper
tantrums, a relatively short attention span, some oppositional behavior, and
aggression. Prolonged temper tantrums, extreme irritability, or pervasive
oppositional behavior would not be expected; additional evaluation and
behavioral supports would be indicated. Determine how these behaviors affect
family functioning and what supports the family has to manage
them.
Maturationalprogress
Pubertal development should be expected within the same age
parameters as for children without Down syndrome.
Social & Family Functioning
The understanding of Down syndrome by parents, siblings, and
extended family members and their adaptation to the child's special needs should
be discussed. When you meet with a family whose child has just been diagnosed,
asking family members if they have known someone with Down syndrome often
uncovers preconceived notions about outcomes. Ask about awareness of community
resources for health care funding (e.g., Medicaid and relevant waivers, caveats
of private insurance, including benefit exclusions and mechanisms to advocate
for appropriate funding), financial supports (e.g., SSI and role of Workforce
Services), services to optimize development and function (e.g., early
intervention, developmental preschool, special education, inclusion models,
private therapies, augmented communication supports), respite, appropriate
recreational/social outlets, and transition (e.g., vocational rehabilitation,
guardianship association). Discuss potential value of one-time or intermittent
family medical leave (FMLA) for the parents. . Current
functional goals, intervention supports, and adaptive equipment should be
reviewed to identify gaps in needed support. Ensure families have access to
information on life and financial planning for their child. Families should
understand the role of a special needs trust to protect funds put aside to
support their child in adulthood; otherwise, they will need to spend down those
funds to obtain/retain Medicaid or SSI funding. Include the child in these
discussions at a developmentally appropriate level. Pubertal development,
self-exploration, menstrual hygiene, and sexuality should be discussed as the
child approaches adolescence. At all ages, ask about
safety concerns, such as taking off in parking lots (consider a DMV form for a
disability parking pass), refusing to wear a seat belt (consider an adapted car
seat), and wandering (consider a tracking device and safety measures to prevent
opening the house doors). Discuss approaches to prevent sexual victimization
(e.g., discussing at the child’s level appropriate/inappropriate touch, teaching
normal sexual function if appropriate for the teens cognitive level, encouraging
parents to ensure all programs the child participates in have policies on
prevention, and having family members treat the teen with appropriate body space
behaviors for age).
Physical Exam
General
In a child with suspected Down syndrome, the presence of minor anomalies should
be documented. Because these may contribute to parents' concern about the
stigma of Down syndrome, reassurance about their presence is important.
Common minor anomalies include:
Upward-slanting eyes
Inner epicanthal folds
Small upturned nose with depressed nasal bridge
A protruding tongue that develops fissures with age
Brushfield spots
Small ears
Short neck with redundant skin folds
Brachycephaly
Flat occiput
Single palmar (simian) crease
Wide space between first and second toes (sandal
ga)
Clinodactyly of the fifth finger
All of these can be found in
individuals without Down syndrome. The presence of multiple such anomalies
raises suspicion for Down syndrome or another genetic syndrome. After
chromosome results are available, the minor anomalies play little role in
health care decisions and mentioning them may result in unnecessary focus on
clinically insignificant physical differences.
Vital Signs
Document baseline vital signs and oxygen saturation: Since Bright Futures does
not recommend BP measure for the typical healthy child until age 3 years,
office staff may need to be guided to specifically check BP at well-child
checks for children with Down syndrome. This is particularly important if
the child has cardiovascular issues.
Growth Parameters
Height, weight, and head circumference (OFC) should be plotted on
typical growth charts. Down syndrome growth charts for US children were
published [Zemel: 2015] and are helpful for height,
weight, and OFC. Nutritional status should also be assessed with weight for
height (under age 2 years) and BMI (over age 2 years). The authors of the
Down syndrome growth charts felt they should not be used for assessment of
BMI as they were impacted by a high rate of obesity in the study population,
rather typical growth charts should be used to assess the appropriateness of
weight for height (WHO curves) and BMI (CDC curves).
Skin
Note dry skin, cheilitis, evidence of skin infection, eczema, thickened skin on
palms or soles, vitiligo and alopecia.
HEENT/Oral
Assess extra-ocular movements, ocular alignment, pupil response, and presence of
nystagmus. Abnormal red reflex or corneal clouding may indicate cataract.
Look for evidence of nasolacrimal duct obstruction or chronic blepharitis.
Look for middle ear effusions, evidence of chronic sinus infection, or poor
nasal flow suggesting adenoidal enlargement. Monitoring for persistent
middle ear fluid is critical, though often very difficult without special
equipment (due to often very narrow ear canals) and clinical signs of
persistent effusion may be minimal. Some clinicians recommend routine
referral to an ENT for optimal monitoring. Tonsillar and adenoidal
hypertrophy may contribute to airway obstruction. Palpate for thyroid
enlargement or nodules.
Chest
Observe for signs of airway obstruction and or chronic lung disease.
Heart
Assess for murmurs, abnormalities in the first and second heart sound, or evidence
of heart failure.
Abdomen
Bloating may be seen in children with celiac disease or chronic constipation.
Hepatomegaly may be seen with congestive heart failure. Due to low tone, a
protuberant abdomen is common. Umbilical hernias and diastasis recti are
also very common.
Genitalia
Many infants with Down syndrome have a suprapubic fat pad that buries the base
of the penis. Many parents will have concerns for micropenis, but in most
cases, compression of this fat pad will reveal a normal phallus. This fat
pad can also create challenges with circumcision if it creates pressure on a
Plastibel or can lead to a circumferential adhesion of redundant penile skin
around the glans after circumcision if parents are not carefully retracting
any redundant tissue on a daily basis during diaper changes. Assess Tanner
stage. Testicular examination during yearly physical exam is important,
particularly in young adults who are unlikely to do
self-exam.
Extremities/Musculoskeletal
Monitor skeletal alignment; individuals with Down syndrome
are at increased risk for scoliosis. Examine for evidence of hip
abnormalities, including dysplasia, slipped capital femoral epiphysis
(SCFE), dislocation, and avascular necrosis of the femoral head (AVN). Pes
planus is common but rarely requires intervention. Observe gait for
asymmetries, hyperextension at the knees, foot inversion or eversion. Many
children have atypical gaits in part due to poor motor control and low tone;
some children with Down syndrome will benefit from
orthotics.
Neurologic Exam
Regular assessment of hypotonia allows for periodic discussion of
developmental progress and prognosis. Children with more extreme hypotonia
may experience slower gross motor progress. Monitor for seizures through
clinical history. Since patients who have experienced atlanto-axial
dislocation generally have had warning signs, it is important to monitor for
signs or symptoms of chronic spinal cord injury. Observe for head tilt or
limitations in neck range of motion that suggest AA instability. The yearly
physical should include examination of reflexes, including the Babinski. A
child with symptoms should have immediate evaluation.
Testing
Sensory Testing
Vision Following a normal routine newborn examination, the AAP counsels
consideration of referral to an ophthalmologist within the 6 months of life.
[Bull: 2022] Follow-up with a pediatric
ophthalmologist, or general ophthalmologist familiar with Down syndrome, should
occur annually for 1- to 5-year-olds, every 2 years for 6- to 13-year-olds, and
every 3 years for 14- to 21-year-olds. Hearing In addition to assessment by history at every well-child visit,
AAP guidelines suggest: [Bull: 2022]
Test by an objective method (e.g., otoacoustic
emissions, brainstem auditory evoked response) at birth.
Repeat assessment "by objective method" or behavioral
screening plus tympanometry every 6 months until normal ear-specific
hearing is confirmed (usually around age 4).
Attempt first behavioral audiogram by 1 year and repeat
annually after normal ear-specific hearing is confirmed.
Objective hearing assessment should be repeated
whenever there is parental concern or evidence of persistent middle ear
effusions.
Tympanometry may be helpful in the cooperative child to
detect normal motility or presence of effusions.
Thyroi-stimulating hormone (TSH) at newborn screen, 6 months, and
annually. Check additional thyroid function tests if the TSH is
abnormal. Some experts recommend measuring thyroxine level (free T4) or
thyroid antibodies routinely along with TSH screen; however, there is no
consensus among international guidelines. In the event of a mildly
elevated TSH, obtain a free T4 (to determine need for treatment) and
measurement of thyroid antibodies may help to identify those with
evolving autoimmune thyroid disease; however, only a minority will
progress to hypothyroidism, and no consensus exists on the need to treat
subclinical hypothyroidism.
Celiac testing (e.g., celiac reflexive panel, which guides testing based
upon age and serum IgA level for optimal sensitivity) if any suggestive
symptoms. While there is lack of expert consensus on whether to
routinely screen children with Down syndrome for celiac disease, the AAP
recommends screening for symptoms that may be related to celiac disease
in children with Down syndrome at yearly visits and perform serologic
screening if symptoms are present. Some expert groups now recommend
one-time testing of HLA-DQ2 and HLA-DQ8 to help exclude those
individuals who are not at risk for celiac disease. [Bull: 2022]
[Pavlovic: 2017]
Complete blood count with differential in the newborn
period to screen for myeloproliferative disorder and polycythemia.
Monitoring for resolution of the myeloproliferation (and continued
intermittent monitoring until 3 years of age even after resolution) with
a CBC is indicated in children who have had transient
myeloproliferation.
Hemoglobin annually. Guidelines recommend yearly
monitoring of hemoglobin and historically asking about risk factors of
iron deficiency. Screen with a CRP/ferritin (or reticulocyte hemoglobin)
yearly if there are any risk factors for iron deficiency or if the
hemoglobin is <11.
Imaging
An echocardiogram should be performed on every newborn
with Down syndrome to exclude a cardiac defect. In children diagnosed
with obstructive sleep apnea, evaluation with an echocardiogram may be
indicated on an intermittent basis to assess for pulmonary hypertension.
Echocardiographic screening has been recommended in the AAP treatment
guidelines for ages 13-21 if "there is a history of increasing fatigue,
shortness of breath, or exertional dyspnea or abnormal physical exam
findings, such as a new murmur or gallop." [Bull: 2022]
Consider a KUB in any newborn with Down syndrome if there is concern for
duodenal atresia (double-bubble sign). Additional assessment with upper
gastrointestinal series (upper GI) and/or barium enema should be
considered to assess anatomy in infants with gastrointestinal
symptoms.
Obtain an "unprepped" barium enema for any concern for
Hirschsprung disease.
Neuroimaging is not routinely indicated but should be
considered in a child with macrocephaly or severe microcephaly beyond
that typically observed in Down syndrome, a child whose development
seems atypical for Down syndrome, any child with seizures or a change in
neurologic functioning or developmental regression, and a child who has
abnormalities on neurologic examination that cannot be attributed to
Down syndrome.
The AAP guidelines do not recommend universal screening
of infants with Down syndrome for renal and urologic abnormalities.
However, any child with Down syndrome and urinary symptoms (e.g., UTI,
difficulty with voiding, unexplained enuresis) should have an evaluation
of the urinary tract.
Although not all sports programs have caught up with
this recommendation, the AAP guidelines do not recommend screening for
upper cervical spine instability unless symptomatic. The normal
atlas-dens interval is less than 3.5 mm in children but may normally
reach 5 mm in children with Down syndrome. If signs or symptoms of
atlantoaxial instability emerge, evaluation should proceed with c-spine
x-rays (neutral position followed by flexion/extension only if no
abnormality is seen), neuroimaging, and consultation with neurosurgeon
or orthopedic surgeon with expertise.
Genetic Testing
Newborns can be screened for trisomy 21 with a fluorescent in situ
hybridization (FISH) test when there is clinical suspicion. If the FISH screen
is positive, it should be confirmed with a complete karyotype. [Bull: 2022] In utero diagnostic testing with CVS or
amniocentesis nears 100% accuracy and distinguishes among the genetic types of
Down syndrome: trisomy 21, translocation, and mosaicism. If a screen or cell
free DNA during gestation was abnormal but in utero diagnostic testing was not
completed, a karyotype should be completed in the newborn period to confirm the
diagnosis and genetic etiology. If cytogenomic microarray (CMA) is performed in
the prenatal or newborn period, a karyotype needs to be performed as well
because CMA does not distinguish the standard trisomy 21 from the translocation type.
Fluorescent in situ hybridization (FISH) testing
usually takes 1-4 days; results are not diagnostic. [Bull: 2022]
A karyotype performed on lymphocytes confirms the
diagnosis and genetic type. e. [Bull: 2022]
Karyotypes may take 10 or more days.
Genetic studies should be subsequently offered to the
parents only when the child has a translocation. Prenatal diagnosis in
future pregnancies, either with chorionic villous sampling at 10-13
weeks or amniocentesis at 15-20 weeks, is usually offered.
Preimplantation testing is also available to screen blastomeres for
aneuploidy and translocations, but the cost is substantial since this
process requires in vitro fertilization and ICSI (intracytoplasmic sperm
injection).
It is critical for the clinician to understand that genetic testing is
expensive and not all insurances will cover it. Some insurances will not
approve a karyotype if the child had a positive cell-free DNA in utero
or a positive FISH. This is because, if clinically the child has Down
syndrome, they do not consider knowing if it is a translocation or
mosaic medically necessary (it does not change treatment, recurrence
risk assessment is medically indicated for the parents but not the
child). In general, no testing should be sent without prior
authorization. This is particularly true on the outpatient side, but
insurances will sometimes deny testing sent inpatient,
too.
Other Testing
Sleep Study Obtain a sleep study by age 4 or sooner if any symptoms of sleep
apnea or other non-behavioral sleep problems are noted, such as restless legs. Modified Barium Swallow (MBSS) Guidelines
recommend evaluating for aspiration with an MBSS in any infant with suggestive
symptoms, including marked hypotonia, slow feeding, choking,
recurrent/persistent respiratory symptoms, or failure to thrive. This will be
particularly helpful if a speech and language pathologist is present during the
study to assess and make recommendations. Electroencephalogram (EEG) An EEG can be obtained if there are concerns for seizures.
Particularly helpful to evaluate developmental pattern/progress, evaluate for
concerns such as autism, recommend services to optimize development and to
evaluate older children with behavioral or learning
concerns.
Depending upon sedation needs and the policies of the echo center, obtaining
the recommended echocardiogram may require referral to a pediatric
cardiologist. Children with cardiac lesions need long-term
follow-up.
If hearing screening is not provided by the early intervention program or
school, referral to audiology will be needed. Children who fail screening or
for whom the parents have concerns about hearing or auditory processing
should be referred to audiology. Children with hearing impairment should be
followed routinely.
Guidelines recommend evaluation by an ophthalmologist by 6 months of age
(sooner if the red reflex or another aspect of the eye examination is
abnormal) and on an ongoing schedule.
May be helpful in diagnosis, evaluating recurrence risk and prenatal testing
options (particularly in the case of translocation-related Down syndrome),
and counseling the family regarding etiology and
outcomes.
May be indicated when a child has neurologic findings that are not commonly
seen (e.g., tremor, nystagmus, severe hypotonia), atypical neurologic
findings (e.g., spasticity, ataxia), seizures, relative microcephaly or
macrocephaly, developmental delays beyond those typically seen, or any
regression in development or neurologic function.
Very helpful if there are concerns about sleep apnea or poor sleep.
Treatment & Management
Overview
Since there is no treatment for the underlying genetic abnormality in Down
syndrome, treatment focuses on managing comorbid conditions and optimizing function
and societal participation.
Pearls & Alerts for Treatment &
Management
Respiratory infections can cause hospitalization after NICU discharge
Respiratory infections, such as pneumonia and bronchiolitis, are second only
to congenital heart disease as causes for hospitalizations and are
associated with increased morbidity and mortality compared to other children
hospitalized for the same infections.
Altitude and pulmonary edema
A case series that included 6 children with Down syndrome reported
high-altitude pulmonary edema. after traveling to moderate altitudes
(1738-3252 feet) for a short time (several in under 24 hours). [Durmowicz: 2001] Among the 6 children, 4 had congenital heart
defects, 3 had chronic pulmonary hypertension, and 5 had developed an
intercurrent illness just before their travel. The author suggested care
when traveling to even moderate altitudes with children with Down syndrome.
Other authors have suggested an increased risk for those with obstructive
sleep apnea perhaps related to altered chemo responsiveness to hypoxia.
[Richalet: 2008]
Atlantoaxial instability
New onset of focal weakness or bowel/bladder incontinence merits urgent
neurosurgical consultation. More of my time is spent these days explaining
why x-rays are not needed to screen, helping them know the signs to watch
for (and that these are rare but important to take seriously), and
encouraging them to have their child participate in sports avoiding the very
few activities that guidelines suggest restricting (head first diving,
trained tumbling (toddlers are going to do it and stopping them just wants
to make them do it more), rugby, trampoline before 6 years of age and after
that ensure supervision).
Polycythemia and sleep apnea
Increased red blood cells may be noted in the setting of chronic sleep apnea
or chronically inadequate fluid intake.
Psychoactive medications
Many children with neurodevelopmental disorders are sensitive to psychoactive
medications. Use very low doses (e.g., half the starting dose for a
neurotypical child) when starting medications, such as SSRIs, and titrate
slowly to avoid activation and other side effects. Having said that, many
children with Down syndrome benefit from careful management of mental health
issues with medications such as stimulants for ADHD, SSRIs for depression or
anxiety, and mood stabilizers for self-injury in the face of ASD. Review
with families carefully potential side effects (e.g., agitation, insomnia)
and targeted symptoms (e.g., anxiety, impulsivity, depression
symptoms).
Anticipatory well care
Safety and toilet training (often as part of the child’s IEP) are often
overlooked in the fray of the child’s medical needs, but they are critical
to include in the child’s preventive care discussions.
How should common problems be managed differently in children with Down Syndrome?
Growth or Weight Gain
The most current guidance is to use a standard growth chart
complemented by the Down syndrome growth charts published in 2015 (and not the
widely distributed Down syndrome growth charts previously
published).
Development (Cognitive, Motor, Language, Social-Emotional)
Be aware that some children with Down syndrome may also have autism spectrum
disorder (ASD). A specialist should perform formal evaluation for an ASD in the
context of Down syndrome. Evaluation can be helpful in accessing services
specific to treatment of ASD, such as Applied Behavior Analysis (ABA therapy).
See Autism Spectrum Disorder for
more details.
Viral Infections
Children with Down syndrome can have significant morbidity from viral
respiratory infections, particularly in the face of a heart defect, pulmonary
hypertension, severe LTM, or significant sleep obstruction. Some children may
qualify for Synagis to reduce the risk of RSV infection; based on age and
comorbidities, influenza vaccination should be encouraged. Many children have
had recurrent croup, and proactive provision of single-dose steroids can be
helpful.
Bacterial Infections
Due to narrow ear canals, it can be challenging to visualize the
tympanic membranes. Sometimes, given the clinical scenario (e.g., the child has
had a URI for a number of days and suddenly becomes febrile and sleeping
poorly), the clinician suspects but cannot tell and may have to make a judgment
call. Over time, work with an ootolaryngologist to determine if persistent
effusions are present and to place tubes for recurrent middle ear infections.
Children with Down syndrome have an increased prevalence of sinusitis. Many
children with Down syndrome will benefit from a Pneumovax vaccination at age 2
years based upon comorbidities.
Other
Many children with Down syndrome have significant oral sensory symptoms.
Consideration of this when prescribing oral medications (e.g., pill crushed vs.
liquid vs. sprinkles, compounding for taste) may help with
compliance.
Systems
Cardiology
Congenital heart defects are found in 44% of infants with Down syndrome.
[Plaiasu: 2017] Children with Down syndrome are
more likely than other children with similar congenital heart defects to
develop increased pulmonary vascular resistance. Fixed pulmonary vascular
obstructive disease can be seen before the first birthday and may present as
an apparent paradoxical improvement in cardiac symptoms. Because of this,
optimal timing of surgical repair differs from similar cardiac lesions in
children without Down syndrome. The primary care clinician should apprise
the child’s cardiologist of any signs or symptoms of airway obstruction or
sleep apnea because these may impact the child’s cardiac management.
Children with Down syndrome and congenital heart
disease should receive all routine childhood immunizations. The importance
of the influenza vaccine should be discussed, and Pneumovax and RSV
prophylaxis may be indicated for some children. Endocarditis prophylaxis
prior to dental procedures will be indicated in select patients. See the
Dental and Oral Health Screening page for guidelines.
Adults with Down syndrome are at increased risk of
valvular dysfunction, arrhythmias, and coronary artery disease. [Lin: 2008]
To identify or manage a sleep disorder or sleep-disordered breathing. A
sleep study can help to identify the type (obstructive, central, or
mixed) and severity of suspected sleep apnea, as well as other sleep
disorders, such as restless leg syndrome.
Nutrition/Growth/Bone
Nutritional monitoring/intervention is critical to prevent
over/undernutrition and to promote self-feeding. See Nutrition under
comorbid conditions for nutritional risk factors. Suboptimal growth is
characteristic and begins during gestation. Growth should be plotted and
followed on the same growth charts used for children without Down syndrome
as well as the growth charts for Down syndrome published in 2015.
[Bull: 2022] See Down Syndrome Growth Charts (CDC). Some
children will need significant support during the first few weeks of life to
attain success with nursing or bottle feeding – these may include
positioning, special nipples, thickening of formula, special feeding
techniques (e.g., chin or jaw support), more frequent feeding, higher
calorie formulas, or supplemental tube feedings. A speech therapist or
occupational therapist can assess the child's suck and make recommendations
regarding feeding technique. A low threshold should be maintained for a
video/modified barium swallow to assess safety and optimal thickness of
liquids. A Board Certified Lactation Consultant may be critically important
in supporting successful breastfeeding. Feeding
therapy may also be important in the second half of infancy if a child has
difficulty accepting new tastes or textures. Some children who have not had
aspiration issues can develop them. So, even if a swallow study has been
performed previously, a low threshold should be maintained to repeat it for
concerns of aspiration. Lack of satiety and compulsive eating frequently
contribute to excessive weight gain. Parents should structure portions and
offer reasonable portions if the child compulsively overeats. A referral for
behavioral support can help a family implement a healthy diet, provide a
structured feeding plan (e.g., the child refuses to eat at mealtimes and
grazes all day), and discuss dietary changes for excessive weight gain or
has food-seeking behaviors. Consider prescribing a standard multivitamin to
ensure adequate vitamin and mineral intake but ensure families understand
that multivitamins do not replace all the micronutrients present in a more
optimal diet. Consider testing for thyroid
dysfunction and celiac disease in children with inadequate linear growth.
However, feeding behavior and dysphagia concerns more frequently cause
failure to thrive than thyroid or celiac disease. Growth hormone markers
should be checked if the growth pattern is suggestive of growth hormone
deficiency (e.g., failed linear growth despite good nutritional
reserves).
Helpful in assessing nutritional status and adequacy of caloric intake,
recommending special formulas and/or nutritional supplements,
determining safety of nutritional supplements used for complementary
therapy, and may guide the treatment of obesity.
May provide evaluation and intervention to optimize communication (verbal
or using augmentative approaches) and cognitive abilities. In some
communities, they take the role of feeding therapists. In some
cases, the speech and language pathologist who is evaluating the
child through early intervention can evaluate the child's feeding
skills.
Can provide intervention focused on feeding, as well as the treatment of
the oral sensory issues that may affect feeding, dietary choices,
behavior, and function. In some cases, the occupational therapist
who is evaluating the child through early intervention can evaluate
the child's feeding skills.
Consultation with a Board Certified Lactation
Consultant is important for the family that desires to breastfeed
their infant with Down syndrome.
Respiratory
Children with Down syndrome are predisposed to pulmonary conditions that can
lead to recurrent acute illness and/or chronic lung disease. For more
detail, see the Respiratory section under Comorbid Conditions in
Down Syndrome above.
Strongly consider administration of the polysaccharide pneumonia vaccine
(23-valent), as early as age 2, and encourage annual influenza vaccination.
RSV prophylaxis may be indicated in some patients. [Watts: 2013]
Management of chronic lung disease is similar to
that in patients without Down syndrome. The Portal's Asthma provides helpful information on the
diagnosis and treatment of asthma/chronic airway inflammation; however,
wheezing in Down syndrome often does not stem from asthma (although children
with Down syndrome can have asthma like any other child!) and therefore its
some cases will be less responsive to bronchodilators and controllers.
Involvement of a pulmonary specialist can be helpful to manage decisions
regarding the use of these. Consider an immunology
evaluation and/or evaluation for Gastroesophageal Reflux Disease and/or oral aspiration in
children with repeated pneumonias, other pyogenic lung infections, or
chronic lung disease. High-altitude pulmonary edema
can occur in children with Down syndrome. Care should be taken when
traveling to moderate altitudes, particularly for those with cardiac
defects, pulmonary hypertension, or obstructive sleep apnea that might
contribute to altered chemoresponsiveness to hypoxia. [Watts: 2013] If parents note symptoms, they should retreat to a lower
threshold and seek care emergently if symptoms persist. If the symptoms
resolve, they should discuss with their clinicians a plan to manage future
travel to higher elevations.
May be helpful for children with recurrent or persistent pulmonary symptoms, chronic lung disease, chronic respiratory symptoms,
recurrent pneumonia, or acute compromise in breathing/air exchange.
Sleep
Sleep apnea occurs in 30-60% of children with Down syndrome and may be
asymptomatic. [Farhood: 2017] See the Comorbid
Condition section in Down Syndrome, above,
for a discussion of the factors contributing to sleep apnea and suggestive
symptoms. Evaluation should include a sleep study (note: nap studies may be
significantly less sensitive than overnight studies). Note that insurance
funding issues and children’s sensory and anxiety issues can sometimes
impact the reality of getting a sleep study. The following may also be indicated:
Echocardiography: If a sleep study is positive for
obstructive sleep apnea, an echo helps to evaluate for pulmonary
hypertension. Over time a child may need a repeat echo if the OSA
persists, particularly if treatment is ineffective or adherence is
poor.
Chest radiography may be considered if there is
concern for comorbid chronic lung disease.
Hemoglobin - chronic hypoxia due to OSA may result
in polycythemia
Serum bicarbonate and/or early morning blood gas
will help determine the extent of carbon dioxide retention. This can
be ordered as part of the sleep study if there is clinical concern
for hypoventilation.
Evaluation by a pulmonologist boarded in sleep
medicine and by an ENT specialist should be considered for all
children with OSA to develop an optimal treatment plan.
Consider evaluation for gastro-esophageal reflux if
there are suggestive symptoms.
Interventions may include:
Adenoidectomy and tonsillectomy are often
successful in improving symptoms, although often symptoms do not
completely resolve and may require additional interventions such as
CPAP.
Postoperative apnea is a frequent
complication, suggesting a need for longer postoperative
monitoring. [Farhood: 2017]
[Nation: 2017]
A follow-up sleep study should be
considered approximately 6-8 weeks after surgery.
A sleep endoscopy may be performed in some
patients to determine anatomic contributors to obstruction,
particularly if tonsils and adenoids do not appear to be the
problem clinically.
Treatment for chronic sinusitis, allergies, or GERD
may be helpful in the child with suggestive symptoms. [Brouillette: 2001]
Nighttime oxygen and/or continuous positive airway
pressure (CPAP) may be recommended. Some patients are treated with
high-flow nasal cannula oxygen, although the role of HFNC vs. CPAP
is yet to be clarified. Patients often do not tolerate these devices
due to oral hypersensitivity, so a behavioral desensitization
program may be necessary. Parents need extensive support as they
face many challenges (sometimes insurmountable) in providing these
treatments, are often frustrated, and are often extensively sleep
deprived. Desensitization techniques can be found at
CPAP & BIPAP Therapy for Children.
A plan for lifestyle intervention should be
discussed with the family If obesity is contributing to OSA.
Other surgical procedures may be indicated when the
above have failed (e.g., uvulopalatoplasty, tongue reduction
surgery, tracheostomy).
Helpful in diagnosing and managing a sleep-disorder or sleep disordered
breathing. A sleep study can help identify sleep apnea and its
cause, as well as other sleep disorders such as restless leg
syndrome.
Indicated in almost all children with documented or suspected obstructive
sleep apnea to determine the contributors such as enlarged tonsils,
adenoids, laryngomalacia.
Behavioral specialists who are comfortable with
children with cognitive challenges may be very helpful in designing
and supporting interventions to improve sleep.
Hematology/Oncology
Obtain a CBC in the newborn to screen for transient myeloproliferative
disorder and polycythemia. Subsequently, monitor hemoglobin annually, with
testing for iron deficiency anemia when there are concerns (e.g., with a
ferritin and CRP, transferrin saturation, or reticulocyte hemoglobin).
Routine monitoring for leukemia is not advised despite the increased
lifetime risk as it is still very rare and presents symptomatically. If the
infant had myeloproliferative disorder, monitor the complete blood count for
several years after resolution. [Bull: 2022]
Gastro-Intestinal & Bowel Function
GI anomalies occur with increased frequency in Down syndrome– see the
Down Syndrome Comorbid Conditions section for detail.
In the newborn period, significant vomiting or failure to pass meconium
warrants immediate evaluation. The infant with significant chronic
constipation should be evaluated for Hirschsprung's disease; the
incidence is 25-fold higher in DS, and there is high mortality
associated with enterocolitis, particularly in those patients with
cardiac malformations. [Ieiri: 2009]
The prevalence of celiac disease in individuals
with Down syndrome is about 4-10% among US Caucasians (compared to
1:150-300 in the general population). About 1/3 of individuals with
Down syndrome and celiac disease have no overt clinical symptoms.
[Pavlovic: 2017] Treatment includes
life-long dietary exclusion of wheat, rye, barley, and possibly
oats; identification and treatment of complications (e.g., anemia,
malnutrition); and possibly evaluation of family members. See the
Medical Home Portal's Celiac Disease for management
details.
Functional gastrointestinal disorders such as
esophageal dysmotility and chronic constipation can be a problem in
children with Down syndrome and can lead to pain and decreased
appetite as well as behavioral problems. [Moore: 2008] See Constipation and Gastroesophageal Reflux Disease for more detailed
management information.
May assist in the evaluation/management of vomiting, constipation, GERD,
dysphagia, poor oral intake, chronic diarrhea, or suspected celiac
disease.
Neurology
See Comorbid Conditions section on neurologic conditions (microcephaly,
seizures, myelopathy). In addition, special consideration should be given to
the following two presentations:
New onset of weakness has a broad differential diagnosis, is likely
serious, and deserves prompt evaluation. [Worley: 2004] Urgent neurology consultation is indicated for
new onset of focal weakness. Due to atlantoaxial instability,
clinicians need to remain vigilant for signs of spinal cord injury
and myelopathy. All families should be educated on the signs and
symptoms of myelopathy (change in gait or use of arms or hands,
change in bowel or bladder function, neck pain, stiff neck, head
tilt, torticollis, how the child positions his or her head, change
in general function, or weakness), how to avoid high-risk activities
like unsupervised trampoline use, football, rugby, heading balls
during soccer, specific gymnastics, and head first diving, and to
seek care if signs/symptoms emerge. Evaluation should proceed with
c-spine X-rays (neutral position followed by flexion/extension only
if no abnormality is seen), neuroimaging, and consultation with a
neurosurgeon or orthopedic surgeon with expertise in this area.
Families should also know about positioning precautions during
anesthesia/sedation, surgical procedures, and radiographic
examinations.
Regression in function: Alzheimer-type dementia is not seen in pediatric
or young adult patients with Down syndrome. However, a small number
of teens will present with a sudden decline in function. The
patients should have a complete evaluation to exclude the issues
listed below. Although rare, autoimmune encephalitis has been
reported in teens with Down syndrome and sudden loss of cognitive
function and should also be excluded if clinically suspected based
on presentation. A few patients will have no explanation and be
diagnosed with Down syndrome Disintegration Disorder and, in case
series, only a subset have regained prior functional level.
[Worley: 2015] Catatonia in adolescents
and young adults with Down syndrome has been described, and several
case series suggest potential therapeutic interventions.
[Ghaziuddin: 2015]
[Miles: 2019]
Hearing problems
Visual deficits
Thyroid disorders
Sleep problems (e.g., sleep apnea can
contribute to impaired attention)
Impaired expressive communication
Education or occupational setting not
appropriate for cognitive level or learning style
Emotional problems (e.g., depression,
anxiety)
Auditory processing disorders
Boredom due to lack of recreation or social
outlets
May be helpful in evaluating unusual developmental patterns (e.g.,
associated autism) or cognitive decline.
Mental Health/Behavior
See Down Syndrome,
Comorbid Conditions section for details about dual diagnosis and
evaluation. Behavioral supports and parent training
remain the best management for behavioral challenges. However, carefully
targeted use of psychotropic medication can be beneficial. Little research
guides medication use for ADHD, depression, anxiety, and cognitive function
in children with Down syndrome. In general, individuals with
neurodevelopmental disabilities may have idiosyncratic reactions to
psychotropic medications. A patient may show a positive response at a low
dose or significant adverse effects at a minimal dose. Initiating treatment
at a low dose (e.g., half of the starting dose for a neurotypical child)
with a gradual upward titration may help identify the appropriate dose for a
patient while minimizing adverse effects. Recognize the lack of evidence
upon which to base treatment, cautiously define for the family the goals of
the medication to be observed (e.g., focus in the case of ADHD), and watch
for side effects. Families need to understand the specific goal of the
medication (i.e., the treatment target) as opposed to expecting a broad
impact on their child’s behavior. The most commonly
prescribed types of psychoactive medication for children with Down syndrome
in the United States include stimulants, selective serotonin reuptake
inhibitors, atypical antipsychotics, and alpha-adrenergic agonists.
[Downes: 2015] A 2016 study of guanfacine used
to treat symptoms of ADHD in children with Down syndrome showed a positive
impact on irritability and hyperactivity, and the medication was generally
well-tolerated. [Capone: 2016]
There has been limited study regarding interventions
for autism spectrum disorders in Down syndrome. A small trial of risperidone
in children with Down syndrome, autism, severe intellectual disability,
disruptive behaviors, and self-injury demonstrated the potential for benefit
but cautioned that side-effects (weight gain, metabolic alterations) might
limit long-term utility. [Capone: 2008] See
Autism Spectrum Disorder for a discussion of behavioral and
educational interventions and medications used in the approach to autism.
A 2017 review of pharmaceutical trials aimed at
treating cognitive and adaptive function impairments in individuals with
Down syndrome revealed many more questions than answers; current definitive
guidance is lacking. [Keeling: 2017]
This category includes all types of counselors/counseling for children.
Once on the page, the search can be narrowed by city or using the
Search within this Category field.
Often includes developmental pediatricians, neurologists, psychologists,
speech and language pathologists, and occupational therapists; can
document current functional abilities and make recommendations for
intervention programming.
Can provide support in ensuring optimal health monitoring, identification
of comorbid conditions, assessing developmental progress and
assuring optimal intervention services, and management of behavioral
concerns.
Behavioral specialists who are comfortable with
children with cognitive challenges may be very helpful in designing
and supporting interventions to improve sleep.
Ears/Hearing
By adulthood, 60-80% of individuals with Down syndrome have hearing loss due
to sensorineural, conductive, or combined causes. During early development,
even minor hearing loss can negatively impact the development of hearing,
speech, and intellect. Although often hard to diagnose, auditory processing
deficits may exist in some children. See Ears/Hearing section above under
Comorbid Conditions above for details of hearing monitoring.
If a child has an auditory/hearing impairment,
consider:
Full evaluation and monitoring by an audiologist
for diagnosi to determine benefit from amplification (e.g., hearing
aid, FM trainer, cochlear implant) and family support.
If diagnosed in the newborn period, consider
sending testing for CMV (in some states, this may be mandated by
law).
All children should be evaluated by an
otolaryngologist. If the hearing loss is conductive, ventilation
tubes may be indicated.
Evaluation by a speech and language therapist for
program planning and family support.
Notification of the child's teacher so that the
child’s IEP can be adapted to provide appropriate classroom
modifications. The public schools hearing support services should be
consulted to advise on classroom modifications such as:
Limiting background noise in teaching
environments
Optimal positioning of the child in the
classroom
Ensuring the child can always see the
speaker's face
Slowing the pace of verbally presented
material
Checking in with the child to verify
understanding
Increased use of visual materials in the
classroom
Total or alternate communication
programming
An intervener or aid indicated
Local programs for the hearing impaired should be
contacted to advise the child's school, teacher, and family.
Vision should be evaluated to ensure there is no
additional sensory deficit.
Once a hearing impairment is identified, continued
monitoring of hearing and vision are indicated to identify any changes over
time. See the Hearing Loss & Deafness for more
information.
Can provide hearing screening, monitor hearing status, evaluate for and
adjust amplification, and help families identify intervention
services and adaptations.
Consider referral for recurrent otitis media and/or conductive hearing
loss, or if unable to visualize the eardrum or monitor for effusion.
May also be indicated for obstructive sleep apnea or recurrent sinus
infection.
May offer specialized classroom settings or consultation with a classroom
teacher regarding modifications to aid the child with auditory
impairment. May also offer infant and parent education
programs.
Eyes/Vision
Individuals with Down syndrome are at risk for ocular abnormalities as
outlined in the Clinical Assessment Comorbid Conditions section. Monitoring
vision is key to preventing secondary, preventable/treatable disabilities.
Management of visual impairment may be complicated by the child's ability to
tolerate glasses, patching, or other intervention. Fitting glasses for
children with Down syndrome can be very challenging due to their differences
in mid-face structure – SPECS4US provides
valuable resources related to eyeglasses.
May offer specialized classroom settings or consultation with a classroom
teacher regarding modifications to aid the child with visual
impairment. May also offer infant and parent education
programs.
Dental
Children and young adults with Down syndrome are at risk for multiple oral
and dental disorders, as outlined in the Down Syndrome,
Comorbid Condition section. To assist in the
prevention and early detection of dental disorders and associated
complications, the primary care provider should:
Encourage routine dental care. Families may need
support in identifying a provider and/or advocating for funding.
Dental check-ups are recommended by age 1 year and then every 6
months. If indicated, facilitate care by offering the dental care
provider relevant information about Down syndrome. See
Dental and Oral Health Screening for more information
about how to assess and prevent dental problems in the primary care
setting.
Provide fluoride varnish at well checks as
recommended by the Bright Futures Guidelines and ensure adequate
home water fluoridation or prescribe supplementation.
Address behaviors related to children resisting brushing due to sensory
issues and feeding behaviors that result in grazing, frequent bottle
sipping, or night time oral intake. Many children are on thickened
water. Unfortunately, thickened water results in frequent
carbohydrate exposure to the teeth and can result in severe caries.
If improvement in dysphagia allows, discontinue thickened water as
soon as possible.
Ensure that families and dental providers are aware of medical issues
that may affect care (e.g., need for bacterial prophylaxis, sedation
risks).
Monitor general oral hygiene and dental health and
discuss issues with families as they arise. If signs of periodontal
disease are evident, refer as soon as possible.
Help the child/teen/family manage halitosis, which may significantly
affect social inclusion. Simple interventions, such as tongue
brushing, mouthwashes, breath fresheners, and better dental hygiene,
may help. Medical issues that can cause halitosis include chronic
sinusitis, gastroesophageal reflux, Drooling in Children with Special Health Care Needs,
and periodontal disease.
It is important that a dentist has previously worked with children with
special health care needs and is equipped to provide safe sedation
for procedures as necessary.
May be more comfortable with children with developmental delays. Referral
to special centers may be necessary if a child requires sedation for
dental treatment, particularly if the child's medical status places
them at increased risk for complications of
sedation.
Patients with missing/malformed teeth, dental crowding, malalignment, or
malocclusion should be referred to an orthodontist familiar with
treating these issues in patients with Down
syndrome.
Funding is often a barrier to optimal dental care. Some programs offer
assistance with dental funding.
Endocrine/Metabolism
Thyroid disorders, particularly congenital, autoimmune, and subclinical
hypothyroidism, occur with increased frequency. Management of hypothyroidism
does not differ from those without Down syndrome; no guidelines have been
established for managing subclinical hypothyroidism in children with Down
syndrome. The medical home might opt to follow a very low elevation of TSH
by checking every 6 months in the older child (such as those less than 12
years with normal free T4). A low threshold for referral is appropriate,
particularly in the younger child (given the brain’s vulnerability), if
growth is not optimal, the child has a goiter or thyroid nodule, thyroid
antibodies are positive, or the parents have a high level of concern.
[Murillo-Vallés: 2020]
[Crisafulli: 2019]
May be helpful in diagnosis and management of thyroid dysfunction or other
hormonal disorders.
Immunology/Infectious Disease
Individuals with Down syndrome are at risk for autoimmune disorders as
outlined under Down Syndrome,
Comorbid Conditions. Management of autoimmune disorders does not differ from
that in individuals without Down syndrome. The Down syndrome screening
guidelines only address routine screening for thyroid disorders and celiac
disease. Clinicians need to stay attuned to signs and symptoms that might
suggest the development of other autoimmune conditions (e.g., Addison's,
type I DM).
Can evaluate children with arthritic symptoms and diagnose and treat
conditions such as juvenile idiopathic arthritis and
lupus.
Musculoskeletal
Individuals with Down syndrome are at risk for primary anatomic skeletal
differences as well as complications of hypotonia, ligament laxity, and
increased joint flexibility. See Down Syndrome,
Comorbid Conditions section for details. Regular exercise and weight control
should be emphasized to reduce the risk of degenerative musculoskeletal
disease. [Mik: 2008] Some authors suggest yearly
monitoring by an orthopedic surgeon for prompt identification and management
of musculoskeletal disorders that may limit function. [Caird: 2006]
Can monitor the
musculoskeletal exam of children at risk, evaluate and optimize
gait, and provide management options for identified musculoskeletal
problems.
Referral may be indicated if cervicospinal instability is identified on
screening X-rays or by symptoms.
Skin & Appearance
Individuals with Down syndrome are at risk for a number of dermatologic
conditions. See the Down Syndrome Comorbid
Conditions section for details of these conditions.
Patients with alopecia areata and/or vitiligo
should be evaluated for other autoimmune conditions, including
thyroid disorders and celiac disease. Therapies may be helpful
(e.g., topical and intralesional steroids), though individual
response varies and there is a high rate of spontaneous remission
and relapse. Psychosocial support, coping mechanisms, and peer
education may be important.
Angular cheilosis may be treated with a mild
steroid cream unless fungal or bacterial super-infection is
suspected.
Syringomas may be removed with laser treatments if
indicated.
A dermatologic consult should be considered if atopic dermatitis, dry
skin, or xerosis is resistant to treatment (which is often the
case).
Thickened skin on palms and soles is common and
benign.
Chronic staph folliculitis is a challenge for some
patients and can be minimized using dilute bleach baths or skin
cleansing regimes using HIbiclens or CLnWash
Helpful for chronic, severe, or recurrent skin infections such as folliculitis, angular chelosis, and Norwegian scabies.
Genito-Urinary
Individuals with Down syndrome are at risk for multiple renal and collecting
system abnormalities as outlined in the Down Syndrome,
Comorbid Conditions section. Treatment of urologic conditions is based upon
the malformation present and should be guided by a pediatric
urologist.
Helpful for patients with urinary tract abnormalities or those with
persistent unexplained urinary symptoms.
Maturation/Sexual/Reproductive
The clinician can provide families and children/adolescents with Down
syndrome with information about these issues as well as model how to talk
about these issues in understandable, accurate ways. Issues of sexuality in
Down syndrome include:
Body parts and pubertal maturation
Personal care and hygiene
Menstrual management
Masturbation
Personal space, privacy, and social norms
The risks of sexual abuse
Dating and marriage
Reproduction and fertility including family
planning and pregnancy outcomes
Sexually transmitted diseases
Individuals with Down syndrome as parents
The clinician should encourage the
family to have open and clear discussions with their child or adolescent so
he or she can learn about what is happening during puberty and understand
the family’s values. The AAP’s 2006 clinical report on Sexuality of Children
and Adolescents with Developmental Disabilities is an excellent resource.
[Murphy: 2006] Also see Patient Education under
Down Syndrome for more
resources for clinicians and families.
A gynecologist with expertise in pediatric/adolescent issues can provide family planning guidance and, when indicated/desired,
menstrual suppression management.
Development/Motor
Gross Motor Impairments Most children with Down syndrome have delayed gross motor
development secondary to hypotonia, ligamentous laxity, decreased muscle
strength, motor planning challenges. Physical therapy is important for young
children with Down syndrome to teach correct strengthening exercises and to
prevent the child from using compensatory motor patterns that will be
detrimental in the long run. Common issues in children who do not receive
adequate physical therapy include::
Standing and walking with their hips in external
rotation, knees stiff, and feet flat and turned out
Sitting with their trunk rounded and pelvis tilted
back
Standing with a lordosis
Before age 3, physical therapy is usually available
through an Early Intervention Program, which will focus on teaching parents
to work with the child in the home. An additional benefit will be the
ongoing education about their child's abilities and how best to work with
him or her. For older children, a physical therapist may design an exercise
program to prevent deconditioning and/or obesity. At school, physical
therapy may design adapted physical education programs.
Fine Motor Impairments Fine motor impairments are common and are usually treated by
working through early intervention in infancy and early childhood, then with
an occupational therapist to improve skills and develop adaptive techniques. Sensory Integration Concerns Many children with Down syndrome are sensitive to touch,
manipulation, and textures in and around their mouth and other parts of
their bodies. Sensory issues may affect oral motor skills, the ability of
the parents to care for the child (e.g., dental and facial hygiene,
feeding), and the willingness/ability of the child to eat a variety of
tastes and textures or to wear certain clothes. Sensory integration issues
may be important in expressive communication since producing communicative
responses requires processing and integrating sensory input. Occupational
therapy and/or feeding therapy are often helpful. Before age three,
occupational and feeding therapy may be available through an early
intervention program, and the focus will be teaching parents to work with
the child in the home. However, not some early intervention programs
consider feeding to be “medical” in nature and will not address these issues
in which case the child will benefit from a private referral. An additional
benefit will be the ongoing education about their child's abilities and how
best to work with him or her. For older children, an occupational therapist
will help with sensory integration issues and fine motor problems. Feeding
therapists are either occupational or speech therapists with additional
training in feeding; they can help with sensory issues around eating and
swallowing. At school, occupational therapy can work with the student on
self-help skills and other means to participate more fully in the academic
program. Oral Motor Impairments Intervention for oral motor impairments may be critical
during early development to facilitate adequate feeding. Subsequent
interventions may be important to facilitate advancement in textures,
improve drooling, and to develop expressive language. A swallow study should
be done early if concerns for aspiration and repeated as skills change over
time or new concerns arise. Feeding therapy may help with tolerating
different foods and textures and safe swallowing. Adaptive skills and transitions Support may be needed to learn general adaptive skills such
as self-feeding, toilet training, self-dressing and bathing. It is critical
to think about what the child is capable of doing for themselves with their
current abilities and how the child can contribute to the household.
Occupational therapists can be very helpful in defining appropriate ADL
goals and helping families break down tasks so the child can learn the
needed steps. Skills such as toilet training may become IEP goals. As
children grow, it is important for them to contribute to the household
(e.g., ability appropriate chores) and interface in their community at the
levels for which they are ready (e.g., playdates, ordering at a restaurant,
buying items at a store, learning to cross streets, participating in
community events and clubs, having recreational outlets). Depending on
interest, ability, and availability, children with Down syndrome may
participate in community-based sports programs or adapted sports. As
individuals become older adolescents, job coaching and job placement
experiences should be part of their EIP. Some individuals with Down syndrome
have attended adapted and traditional tertiary education programs.
For children ages 3-21 years PT, OT, and speech-related services are provided to support mobility, fine motor and self-help
skills, and communication for education-related goals, but not necessarily for medical goals (e.g., to enhance range of motion
or improve feeding skills).
Development/Language
All children with Down syndrome have language deficits and will benefit from
early referral for speech and language therapy. Children experience delays
in both receptive and expressive speech. Often expressive language is
substantially more delayed than receptive language. This is important to
recognize as these children will benefit from alternative approaches to
communication (e.g., sign). Speech disorders such as dysfluency, verbal
apraxia, and articulation disorders are common. Up to 30% of children with
Down syndrome will have autism; however, it is important not to assume that
a child with Down syndrome who is not speaking has autism as there are many
other reasons to have challenges with verbal output (e.g., verbal
dyspraxia). Sensory integration issues factor into expressive communication,
since producing communicative responses requires processing and integrating
sensory input. Oral motor skills play a role in language development (see
Development/Motor above). Intervention to provide appropriate stimuli and a
bridge to verbal communication (alternative communication) is extremely
important. Most children will require language support throughout their
education. The primary care clinician can:
Refer promptly to early intervention service
providers.
Ensure appropriate hearing screening throughout
childhood.
Help families understand the role of alternative
communication methods. While 95% of children with Down syndrome will
ultimately use verbal language, language acquisition is universally
delayed. Alternative communication methods can help until this
occurs and does not retard verbal language development. As soon as
the child is able to produce words with efficiency, the child will
prefer verbal communication.
Screen for signs and symptoms of autism and refer
for additional evaluation if concern.
Work with the family around behavioral issues that
emerge due to limited communication and sensory aversions.
Assist in finding alternative supports when the
early intervention or educational system is unable to meet the
child's needs. Funding through private insurance, Medicaid, or other
community programs may be available. Private referral may be
appropriate for less “educational” goals (e.g., to work on a feeding
disorder, drooling, or oral aversion), since school-based services
must relate to educational goals.
May include developmental pediatricians, neurologists, psychologists, speech and language pathologists, and occupational therapists.
Can document current functional abilities and recommend optimal intervention programming.
Can provide hearing screening, monitor hearing status, evaluate for and adjust amplification, and help families identify appropriate
intervention services and adaptations.
Above age 3 years, most children with Down syndrome will qualify for
services through the special education program offered by their
local school district.
Complementary & Alternative Medicine
Many families of children with special health care needs use complementary
and alternative medicine. Be sure to ask about use of over-the-counter
medications, herbs, nutritional supplements, and homeopathic remedies.
Explain the risks of using unregulated substances, including various types
of unregulated hormones. Identify possible interactions with prescription
medications used by the child. Teaching the family a decision-making process
when they are considering use of an alternative treatment will be helpful
for them as they face options repeatedly throughout the child’s life. See
Complementary and Alternative Medicine (CAM) and
Integrative Medicine for CYSHCN. Chiropractic adjustments could be fatal if
atlanto-axial instability is present. [La: 1990]
There do not appear to be evidence-based protocols for chiropractic
manipulation of patients with Down syndrome. Caution families who are
considering chiropractic manipulation for their child.
Ask the Specialist
The parents are overwhelmed with the child’s behavior - how do I
help?
Contrary to some popular beliefs, many children with Down syndrome have very
challenging behavior. Delays in development, anxiety, oppositional
personalities, communication deficits, short attention span, limited impulse
control, sensory concerns, and autism can be contributors. Start by trying to
understand where the child functions (Are they 4 years old but functioning like
a 2-year-old? Is this just the "terrible twos?") Ask the parent to describe
specific events, which helps to give an understanding of contributing factors.
Sometimes the child’s therapists or teachers can give insight. (Can the child
settle and focus during therapy? Are they acting out in the classroom and if so,
what are the triggers?) If it is just the terrible twos in an older child, talk
about that. If the child is primarily oppositional, parent training in
behavioral techniques is helpful. If there are attention and impulse control
issues or anxiety, depending on the severity, environmental supports, parent
training for behavioral approaches, and sometimes medications are used. In some
cases, refer for an evaluation for autism.
Council on Genetics (AAP) Supports the integration of genetic and genomic medicine in pediatric health care by expanding the genetic literacy of pediatric
teams and supporting the professional needs of geneticists; American Academy of Pediatrics.
Trisomy 21 (OMIM) Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man, authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine
Baumer N, Davidson EJ. Supporting a happy, healthy adolescence for young people with Down syndrome and other intellectual disabilities: recommendations
for clinicians. Curr Opin Pediatr.
2014;26(4):428-34.
PubMed abstract
Watts R, Vyas H. An overview of respiratory problems in children with Down's syndrome. Arch Dis Child.
2013;98(10):812-7.
PubMed abstract
Skotko BG, Capone GT, Kishnani PS. Postnatal diagnosis of Down syndrome: synthesis of the evidence on how best to deliver the news. Pediatrics.
2009;124(4):e751-8.
PubMed abstract
Bull MJ. Down Syndrome. N Engl J Med.
2020;382(24):2344-2352.
PubMed abstract
Korlimarla A, Hart SJ, Spridigliozzi GA, Kishnani PS. Cassidy and Allanson’s Management of Genetic Syndromes. 4 ed. Hoboken, NJ: John Wiley & Sons;
2021.
Clinical Tools
Clinical Checklists & Visit Tools
Down Syndrome Checklist (2022) ( 487 KB) A checklist for recommended monitoring and screening of children with Down syndrome, from the AAP Clinical Report by Bull
et al. (PEDIATRICS Volume 149, number 5, May 2022)
Growth/BMI Charts
Down Syndrome Growth Charts (CDC) Growth charts used on over 1500 measurements on 637 individual with DS, published by Zemel, et al. in 2015, from the Centers
for Disease Control and Prevention's site.
Patient Education & Instructions
Children with Down Syndrome: Health Care Information for Families (AAP) Comprehensive guide to help parents and families of children with Down syndrome. Focuses on medical topics, by age, that affect
physical health. Includes links to health care information for families of children with Down syndrome. PDF downloads available;
American Academy of Pediatrics.
Living with Down Syndrome (Down Syndrome Educational Trust) ( 951 KB) A 26-page, printable booklet with information about family, school, social, and medical issues particular to those with Down
syndrome. It has a positive focus and includes personal stories and helpful advice for parents and families.
A Parent's Guide to Puberty for Children with Disabilities (LEND) ( 7 KB) Toolkits for parents to help adolescents with disabilities learn about puberty, personal hygiene, acceptable public behavior,
and peer relations. Offers versions for girls and boys with disabilities and some translations; Vanderbilt Leadership Education
in Neurodevelopmental Disabilities.
Down Syndrome (MedlinePlus) Excellent, detailed review of condition for patients and families; National Library of Medicine and National Institutes of
Health.
Down Syndrome - Health Issues Site developed and edited/authored by a pediatrician, Len Leshin, MD, who has a son with Down syndrome. Includes a number
of essays by experts about specific health topics and provides other useful links.
Sexuality and People with Disabilities ( 257 KB) This Medical Home newsletter provides information for primary care providers and families including Sexuality and People with
Disabilities; American Academy of Pediatrics recommendations for education about sexuality; tips for parents; and resources,
books, and websites for parents and providers.
Dental Care for the Patient with Down Syndrome Topics covered: Medical problems associated with Down syndrome that can affect dental treatment; proper home care and prevention
of dental disease; techniques to help children with Down syndrome become cooperative dental patients; choosing the right dentist;
and how to communicate effectively with the dental staff. by Dr. Elizabeth S. Pilcher, 1997, but still relevant.
Down Syndrome - Autism Connection Organization focused on providing education, support, and resources related to the unique challenges caused by co-occurring
Down syndrome and autism.
National & Local Support
National Down Syndrome Congress The NDSC, a membership organization, offers parent resources, including a "new parent package" of information, resources for
adult siblings caring for a loved one, and information for people with Down syndrome themselves.
Down Syndrome - Autism Connection Organization focused on providing education, support, and resources related to the unique challenges caused by co-occurring
Down syndrome and autism.
National Down Syndrome Society (NDSS) Human rights organization for all individuals with Down syndrome that focuses on 3 key areas of programming: Resources & Support,
Policy & Advocacy, and Community Engagement.
DS-Connect: The Down Syndrome Registry (NIH) This site offers patients and families opportunities to connect with researchers and health care providers, and access research
data. Patients and families can take confidential health-related surveys and express interest in participating in clinical
studies on Down syndrome. Developed by the Down Syndrome Consortium, led by the National Institutes of Health and involving
several Down syndrome advocacy and professional organizations.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization
or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited
to web-based services, provider locator services, and organizations that serve children from across the nation.
Authors & Reviewers
Initial publication: July 2013; last update/revision: February 2022
Alexander M, Ding Y, Foskett N, Petri H, Wandel C, Khwaja O. Population prevalence of Down's syndrome in the United Kingdom. J Intellect Disabil Res.
2016;60(9):874-8.
PubMed abstract
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics; Committee on Genetics; Society
for Maternal–Fetal Medicine. Practice Bulletin No. 162: Prenatal Diagnostic Testing for Genetic Disorders. Obstet Gynecol.
2016;127(5):e108-22.
PubMed abstract
Aumonier ME, Cunningham CC. Breast feeding in infants with Down's syndrome. Child Care Health Dev.
1983;9(5):247-55.
PubMed abstract
Badeau M, Lindsay C, Blais J, Nshimyumukiza L, Takwoingi Y, Langlois S, Légaré F, Giguère Y, Turgeon AF, Witteman W, Rousseau
F. Genomics-based non-invasive prenatal testing for detection of fetal chromosomal aneuploidy in pregnant women. Cochrane Database Syst Rev.
2017;11:CD011767.
PubMed abstract / Full Text
Bagić I, Verzak Z, Cuković-Cavka S, Brkić H, Susić M. Periodontal conditions in individuals with Down's syndrome. Coll Antropol.
2003;27 Suppl 2:75-82.
PubMed abstract
Barca D, Tarta-Arsene O, Dica A, Iliescu C, Budisteanu M, Motoescu C, Butoianu N, Craiu D. Intellectual disability and epilepsy in down syndrome. Maedica (Buchar).
2014;9(4):344-50.
PubMed abstract / Full Text
Baumer N, Davidson EJ. Supporting a happy, healthy adolescence for young people with Down syndrome and other intellectual disabilities: recommendations
for clinicians. Curr Opin Pediatr.
2014;26(4):428-34.
PubMed abstract
Bertapelli F, Pitetti K, Agiovlasitis S, Guerra-Junior G. Overweight and obesity in children and adolescents with Down syndrome-prevalence, determinants, consequences, and interventions:
A literature review. Res Dev Disabil.
2016;57:181-92.
PubMed abstract
Bocian AB, Wasserman RC, Slora EJ, Kessel D, Miller RS. Size and age-sex distribution of pediatric practice: a study from Pediatric Research in Office Settings. Arch Pediatr Adolesc Med.
1999;153(1):9-14.
PubMed abstract
Boëchat MC, Silva KS, Llerena JC Jr, Boëchat PR. Cholelithiasis and biliary sludge in Downs syndrome patients. Sao Paulo Med J.
2007;125(6):329-32.
PubMed abstract
Book L, Hart A, Black J, Feolo M, Zone JJ, Neuhausen SL. Prevalence and clinical characteristics of celiac disease in Downs syndrome in a US study. Am J Med Genet.
2001;98(1):70-4.
PubMed abstract
Brouillette RT, Manoukian JJ, Ducharme FM, Oudjhane K, Earle LG, Ladan S, Morielli A. Efficacy of fluticasone nasal spray for pediatric obstructive sleep apnea. J Pediatr.
2001;138(6):838-44.
PubMed abstract
Bull MJ. Down Syndrome. N Engl J Med.
2020;382(24):2344-2352.
PubMed abstract
Bull MJ, Trotter T, Santoro SL, Christensen C, Grout RW, Burke LW, Berry SA, Geleske TA, Holm I, Hopkin RJ, Introne WJ, Lyons
MJ, Monteil DC, Scheuerle A, Stoler JM, Vergano SA, Chen E, Hamid R, Downs SM, Grout RW, Cunniff C, Parisi MA, Ralston SJ,
Scott JA, Shapira SK, Spire P. Health Supervision for Children and Adolescents With Down Syndrome. Pediatrics.
2022;149(5).
PubMed abstract / Full Text
Caird MS, Wills BP, Dormans JP. Down syndrome in children: the role of the orthopaedic surgeon. J Am Acad Orthop Surg.
2006;14(11):610-9.
PubMed abstract
Capone G, Goyal P, Ares W, Lannigan E. Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. Am J Med Genet C Semin Med Genet.
2006;142C(3):158-72.
PubMed abstract
Capone GT, Brecher L, Bay M. Guanfacine Use in Children With Down Syndrome and Comorbid Attention-Deficit Hyperactivity Disorder (ADHD) With Disruptive
Behaviors. J Child Neurol.
2016;31(8):957-64.
PubMed abstract
Capone GT, Goyal P, Grados M, Smith B, Kammann H. Risperidone use in children with Down syndrome, severe intellectual disability, and comorbid autistic spectrum disorders:
a naturalistic study. J Dev Behav Pediatr.
2008;29(2):106-16.
PubMed abstract
Committee on Practice and Ambulatory Medicine, Bright Futures Periodicity Schedule Workgroup. 2017 Recommendations for preventive pediatric health care. Pediatrics.
2017;139(4).
PubMed abstract / Full Text Includes a link to the Periodicity Schedule, https://www.aap.org/en-us/Documents/periodicity_schedule.pdf.
Committee on Practice Bulletins—Obstetrics, Committee on Genetics, and the Society for Maternal-Fetal Medicine. Practice Bulletin No. 163: Screening for Fetal Aneuploidy. Obstet Gynecol.
2016;127(5):e123-37.
PubMed abstract
Crisafulli G, Aversa T, Zirilli G, Pajno GB, Corica D, De Luca F, Wasniewska M. Subclinical Hypothyroidism in Children: When a Replacement Hormonal Treatment Might Be Advisable. Front Endocrinol (Lausanne).
2019;10:109.
PubMed abstract / Full Text
de Graaf G, Buckley F, Skotko BG. Estimation of the number of people with Down syndrome in the United States. Genet Med.
2017;19(4):439-447.
PubMed abstract
Dixon N, Kishnani PS, Zimmerman S. Clinical manifestations of hematologic and oncologic disorders in patients with Down syndrome. Am J Med Genet C Semin Med Genet.
2006;142C(3):149-57.
PubMed abstract
Dixon NE, Crissman BG, Smith PB, Zimmerman SA, Worley G, Kishnani PS. Prevalence of iron deficiency in children with Down syndrome. J Pediatr.
2010;157(6):967-971.e1.
PubMed abstract / Full Text
Downes A, Anixt JS, Esbensen AJ, Wiley S, Meinzen-Derr J. Psychotropic Medication Use in Children and Adolescents With Down Syndrome. J Dev Behav Pediatr.
2015;36(8):613-9.
PubMed abstract
Durmowicz AG. Pulmonary edema in 6 children with Down syndrome during travel to moderate altitudes. Pediatrics.
2001;108(2):443-7.
PubMed abstract of particular relevance perhaps in Utah.
Dykens EM. Psychiatric and behavioral disorders in persons with Down syndrome. Ment Retard Dev Disabil Res Rev.
2007;13(3):272-8.
PubMed abstract
Farhood Z, Isley JW, Ong AA, Nguyen SA, Camilon TJ, LaRosa AC, White DR. Adenotonsillectomy outcomes in patients with Down syndrome and obstructive sleep apnea. Laryngoscope.
2017;127(6):1465-1470.
PubMed abstract
Ghaziuddin N, Nassiri A, Miles JH. Catatonia in Down syndrome; a treatable cause of regression. Neuropsychiatr Dis Treat.
2015;11:941-9.
PubMed abstract / Full Text
Gillespie KM, Dix RJ, Williams AJ, Newton R, Robinson ZF, Bingley PJ, Gale EA, Shield JP. Islet autoimmunity in children with Down's syndrome. Diabetes.
2006;55(11):3185-8.
PubMed abstract
Ieiri S, Higashi M, Teshiba R, Saeki I, Esumi G, Akiyoshi J, Nakatsuji T, Taguchi T. Clinical features of Hirschsprung's disease associated with Down syndrome: a 30-year retrospective nationwide survey in Japan. J Pediatr Surg.
2009;44(12):2347-51.
PubMed abstract
Jackson JM, Crider KS, Cragan JD, Rasmussen SA, Olney RS. Frequency of prenatal cytogenetic diagnosis and pregnancy outcomes by maternal race-ethnicity, and the effect on the prevalence
of trisomy 21, Metropolitan Atlanta, 1996-2005. Am J Med Genet A.
2014;164A(1):70-6.
PubMed abstract / Full Text
Juj H, Emery H. The arthropathy of Down syndrome: an underdiagnosed and under-recognized condition. J Pediatr.
2009;154(2):234-8.
PubMed abstract
Keeling LA, Spiridigliozzi GA, Hart SJ, Baker JA, Jones HN, Kishnani PS. Challenges in measuring the effects of pharmacological interventions on cognitive and adaptive functioning in individuals
with Down syndrome: A systematic review. Am J Med Genet A.
2017;173(11):3058-3066.
PubMed abstract
Korlimarla A, Hart SJ, Spridigliozzi GA, Kishnani PS. Cassidy and Allanson’s Management of Genetic Syndromes. 4 ed. Hoboken, NJ: John Wiley & Sons;
2021.
La Francis ME. A chiropractic perspective on atlantoaxial instability in Down's syndrome. J Manipulative Physiol Ther.
1990;13(3):157-60.
PubMed abstract
Lin AE, Basson CT, Goldmuntz E, Magoulas PL, McDermott DA, McDonald-McGinn DM, McPherson E, Morris CA, Noonan J, Nowak C,
Pierpont ME, Pyeritz RE, Rope AF, Zackai E, Pober BR. Adults with genetic syndromes and cardiovascular abnormalities: clinical history and management. Genet Med.
2008;10(7):469-94.
PubMed abstract / Full Text
Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res.
2012;197:101-21.
PubMed abstract / Full Text
Macchini F, Leva E, Torricelli M, Valadè A. Treating acid reflux disease in patients with Down syndrome: pharmacological and physiological approaches. Clin Exp Gastroenterol.
2011;4:19-22.
PubMed abstract / Full Text
Mai CT, Kucik JE, Isenburg J, Feldkamp ML, Marengo LK, Bugenske EM, Thorpe PG, Jackson JM, Correa A, Rickard R, Alverson CJ,
Kirby RS. Selected birth defects data from population-based birth defects surveillance programs in the United States, 2006 to 2010:
featuring trisomy conditions. Birth Defects Res A Clin Mol Teratol.
2013;97(11):709-25.
PubMed abstract / Full Text
Menéndez M. Down syndrome, Alzheimer's disease and seizures. Brain Dev.
2005;27(4):246-52.
PubMed abstract
Mik G, Gholve PA, Scher DM, Widmann RF, Green DW. Down syndrome: orthopedic issues. Curr Opin Pediatr.
2008;20(1):30-6.
PubMed abstract
Miles JH, Takahashi N, Muckerman J, Nowell KP, Ithman M. Catatonia in Down syndrome: systematic approach to diagnosis, treatment and outcome assessment based on a case series of seven
patients. Neuropsychiatr Dis Treat.
2019;15:2723-2741.
PubMed abstract / Full Text
Moore SW. Down syndrome and the enteric nervous system. Pediatr Surg Int.
2008;24(8):873-83.
PubMed abstract
Morris JK, Alberman E. Trends in Down's syndrome live births and antenatal diagnoses in England and Wales from 1989 to 2008: analysis of data from
the National Down Syndrome Cytogenetic Register. BMJ.
2009;339:b3794.
PubMed abstract / Full Text
Murillo-Vallés M, Martinez S, Aguilar-Riera C, Garcia-Martin MA, Bel-Comós J, Ybern MLG. Subclinical hypothyroidism in childhood, treatment or only follow-up?. BMC Pediatr.
2020;20(1):282.
PubMed abstract / Full Text
Murphy NA, Elias ER. Sexuality of children and adolescents with developmental disabilities. Pediatrics.
2006;118(1):398-403.
PubMed abstract / Full Text This American Academy of Pediatrics' Clinical Report provides additional information and guidance for providers relating to
puberty, psychosocial considerations, sexual abuse, sexuality education, and the pediatrician's role.
Nation J, Brigger M. The Efficacy of Adenotonsillectomy for Obstructive Sleep Apnea in Children with Down Syndrome: A Systematic Review. Otolaryngol Head Neck Surg.
2017;157(3):401-408.
PubMed abstract
Natoli JL, Ackerman DL, McDermott S, Edwards JG. Prenatal diagnosis of Down syndrome: a systematic review of termination rates (1995-2011). Prenat Diagn.
2012;32(2):142-53.
PubMed abstract
Oxelgren UW, Myrelid Å, Annerén G, Ekstam B, Göransson C, Holmbom A, Isaksson A, Åberg M, Gustafsson J, Fernell E. Prevalence of autism and attention-deficit-hyperactivity disorder in Down syndrome: a population-based study. Dev Med Child Neurol.
2017;59(3):276-283.
PubMed abstract
Pavlovic M, Berenji K, Bukurov M. Screening of celiac disease in Down syndrome - Old and new dilemmas. World J Clin Cases.
2017;5(7):264-269.
PubMed abstract / Full Text
Plaiasu V. Down Syndrome - Genetics and Cardiogenetics. Maedica (Buchar).
2017;12(3):208-213.
PubMed abstract / Full Text
Richalet JP, Chenivesse C, Larmignat P, Meille L. High altitude pulmonary edema, down syndrome, and obstructive sleep apneas. High Alt Med Biol.
2008;9(2):179-81.
PubMed abstract
Skotko BG, Capone GT, Kishnani PS. Postnatal diagnosis of Down syndrome: synthesis of the evidence on how best to deliver the news. Pediatrics.
2009;124(4):e751-8.
PubMed abstract
Skotko BG, Kishnani PS, Capone GT. Prenatal diagnosis of Down syndrome: how best to deliver the news. Am J Med Genet A.
2009;149A(11):2361-7.
PubMed abstract
Taşdemir HA, Cetinkaya MC, Polat C, Belet U, Kalayci AG, Akbaş S. Gallbladder motility in children with Down syndrome. J Pediatr Gastroenterol Nutr.
2004;39(2):187-91.
PubMed abstract
Unachak K, Tanpaiboon P, Pongprot Y, Sittivangkul R, Silvilairat S, Dejkhamron P, Sudasna J. Thyroid functions in children with Down's syndrome. J Med Assoc Thai.
2008;91(1):56-61.
PubMed abstract
Viegelmann G, Low Y, Sriram B, Chu HP. Achalasia and Down syndrome: a unique association not to be missed. Singapore Med J.
2014;55(7):e107-8.
PubMed abstract / Full Text
Walker JC, Dosen A, Buitelaar JK, Janzing JG. Depression in Down syndrome: a review of the literature. Res Dev Disabil.
2011;32(5):1432-40.
PubMed abstract
Watts R, Vyas H. An overview of respiratory problems in children with Down's syndrome. Arch Dis Child.
2013;98(10):812-7.
PubMed abstract
Worley G, Crissman BG, Cadogan E, Milleson C, Adkins DW, Kishnani PS. Down Syndrome Disintegrative Disorder: New-Onset Autistic Regression, Dementia, and Insomnia in Older Children and Adolescents
With Down Syndrome. J Child Neurol.
2015;30(9):1147-52.
PubMed abstract
Worley G, Shbarou R, Heffner AN, Belsito KM, Capone GT, Kishnani PS. New onset focal weakness in children with Down syndrome. Am J Med Genet A.
2004;128A(1):15-8.
PubMed abstract / Full Text
Zemel BS, Pipan M, Stallings VA, Hall W, Schadt K, Freedman DS, Thorpe P. Growth Charts for Children With Down Syndrome in the United States. Pediatrics.
2015;136(5):e1204-11.
PubMed abstract / Full Text

 Get More Help in Nevada
Get More Help in Nevada
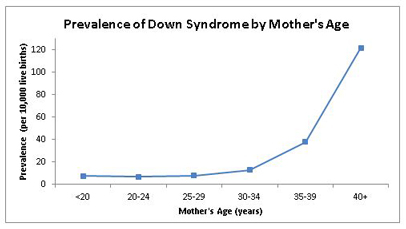
 487 KB))
487 KB))