
 Get More Help in Nevada
Get More Help in NevadaSickle Cell Disease
Overview
Amino Acid Sequence Ultimately Causing Sickle Cells
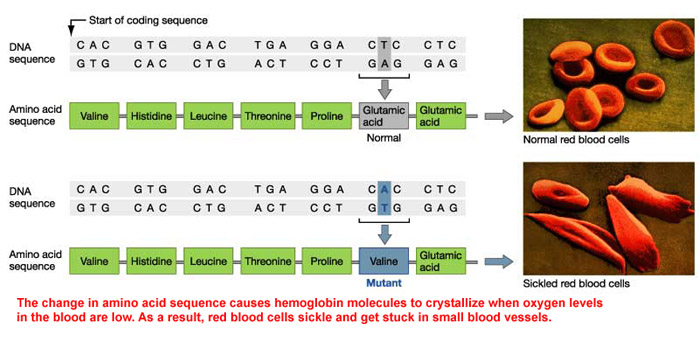
Other Names & Coding
D57.0x, Hb SS disease with crisis
D57.1, Sickle cell disease without crisis
D57.2xx, Sickle cell/Hb-C disease
D57.4xx, Sickle cell thalassemia
D57.8xx, Other sickle cell disorders
An "x" indicates the need or potential for additional digits to provide a more specific diagnosis. More coding details can be found at ICD-10 for Sickle Cell Disorders (icd10data.com).
Prevalence
Genetics
Prognosis
Practice Guidelines
Brandow AM, Carroll CP, Creary S, Edwards-Elliott R, Glassberg J, Hurley RW, Kutlar A, Seisa M, Stinson J, Strouse JJ, Yusuf
F, Zempsky W, Lang E.
American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain.
Blood Adv.
2020;4(12):2656-2701.
PubMed abstract / Full Text
DeBaun MR, Jordan LC, King AA, Schatz J, Vichinsky E, Fox CK, McKinstry RC, Telfer P, Kraut MA, Daraz L, Kirkham FJ, Murad
MH.
American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular
disease in children and adults.
Blood Adv.
2020;4(8):1554-1588.
PubMed abstract / Full Text
Liem RI, Lanzkron S, D Coates T, DeCastro L, Desai AA, Ataga KI, Cohen RT, Haynes J, Osunkwo I, Lebensburger JD, Lash JP,
Wun T, Verhovsek M, Ontala E, Blaylark R, Alahdab F, Katabi A, Mustafa RA.
American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease.
Blood Adv.
2019;3(23):3867-3897.
PubMed abstract / Full Text
Chou S, Alsawas M, Fasano R, Field J, Hendrickson J, Howard J, Kameka M, Kwiatkowski J, Pirenne F, Shi P, Stowell S, Thein
S, Westhoff C, Wong T, Akl E.
American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support.
Blood Adv..
2020;4(2):327-355.
/ Full Text
Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe
PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J.
Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members.
JAMA.
2014;312(10):1033-48.
PubMed abstract
Roles of the Medical Home
Comprehensive sickle cell care is multidisciplinary, and the medical home plays an important role in coordinating this care. With evolving medications to improve the well-being of children with sickle cell disease and possibly cure the disease (see Treatment & Management), the medical home can help families be aware of options and their pros and cons. Primary care clinicians should collaborate with sickle cell specialists and programs to facilitate access to the latest information and best care. Further, primary care can help ensure ongoing engagement and follow-up with a sickle cell specialist to ensure timely preventative assessments such as annual eye exams and stroke screening. A recent study examined the information sources and perspectives of parents and other primary caregivers of children with sickle cell disease related to new therapies.
Clinical Assessment
Overview
Pearls & Alerts for Assessment
Pulse oximetry can underestimate oxygen saturationPulse oximetry is an inaccurate measurement and often underestimates oxygen saturation due to increased levels of carboxyhemoglobin and methemoglobin in patients with sickle cell disease.
Assessment of painChildren experiencing an acute vaso-occlusive pain crisis may not have changes in vital signs (tachycardia or hypertension). Assessment of pain should be based primarily on the patient’s subjective report. Patients may have a higher opioid tolerance over time.
Screening
For the Condition
Of Family Members
For Complications
Stroke risk: Measurement of cerebral blood flow velocities by transcutaneous Doppler ultrasonography should be conducted by those trained in the technique specific for sickle cell anemia. Begin annual testing at age 2 and until age 16 to screen for elevated stroke risk requiring intervention with chronic transfusion therapy.
Silent cerebral infarcts: New guidelines recommend a baseline MRI/MRA without sedation (when tolerable) to evaluate for silent cerebral infarcts and CNS vasculopathy once in early childhood.
Pulmonary hypertension: Routine screening echocardiograms are no longer recommended in asymptomatic individuals; however, those with symptoms or signs of fatigue, exercise intolerance, chest pain, or peripheral edema should be referred for an echocardiogram.
Iron overload: Patients receiving frequent transfusions as well as those on chronic transfusion therapy are at risk for iron overload and should have screening ferritin levels to evaluate the potential for iron overload. If ferritin >1000, screening MRIs for liver iron content should be performed and monitored in conjunction with initiation of iron chelation therapy. [Coates: 2017]
Presentations
The presentation of complications related to sickle cell disease depends on the organ system involved. Those with stroke will usually present acutely with a focal neurologic deficit, such as hemiparesis, aphasia, or ataxia, depending on the anatomic location of the stroke. Depending on the severity or stage of sickle retinopathy, patients may present with spots in the field of vision (floaters), blurriness, decreased visual acuity, or visual field loss. Patients with pulmonary hypertension may present with exertional dyspnea or persistent hypoxia.
Diagnostic Criteria
Clinical Classification
Common Sickle Cell Genotypes
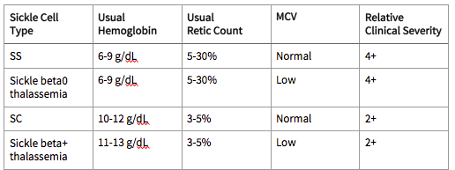
Differential Diagnosis
- Homozygous sickle cell anemia (Hb SS)
- Sickle-Hb C disease
- Sickle β0 thalassemia
- Sickle β+ thalassemia
- Sickle-Hb D disease
- Sickle-Hb E disease
- Other sickle syndromes
Comorbid & Secondary Conditions
- Asthma: Patients with uncontrolled asthma may have more frequent vaso-occlusive pain due to increased sickling with hypoxia.
- Gall stones/hyperbilirubinemia
- Sickle lung disease: Due to chronic sickling within the lung vasculature, patients develop obstructive, and then restrictive, changes in the lungs, which frequently leads to pulmonary hypertension and is the leading cause of death in adults with sickle cell disease.
- Stroke: Children with prior stroke may have residual neurologic deficits.
- Sickle nephropathy: Children with sickle cell disease can develop renal complications over time. This usually starts with urine concentrating defects (hyposthenuria) followed by glomerular hyperfiltration, proteinuria, and eventually focal segmental glomerulonephritis.
- Retinopathy: If untreated, sickling within the retinal vessels can cause proliferative retinopathy, hemorrhage within the retina, retinal detachment, and blindness.
- Osteonecrosis: Infarction of the bones can cause chronic severe pain and limitation in mobility.
- Leg ulcers [Minniti: 2010]
- Priapism
History & Examination
Current & Past Medical History
Family History
Pregnancy/Perinatal History
Developmental & Educational Progress
Maturationalprogress
Social & Family Functioning
Physical Exam
Patients experiencing acute vaso-occlusive pain crises typically appear distressed and very uncomfortable. However, some patients, especially those with frequent pain episodes or chronic pain, may be more stoic or not show the “expected” behaviors of someone with severe pain. The vital signs may be normal. Swelling does not have to be present. Further, there are no radiographic or laboratory findings/dx of a “crisis.”
Vital Signs
Growth Parameters
Children have a decreased growth velocity in proportion to the severity of their anemia.
Skin
HEENT/Oral
Adenotonsillar hypertrophy is the most common risk factor for obstructive sleep apnea in children with sickle cell disease. [Rosen: 2014] In patients with sickle cell disease, obstructive sleep apnea is associated with increased frequency of vaso-occlusive pain crises due to more frequent deoxygenation, which triggers red cell sickling. Children and adolescents with sickle cell disease have an increased risk of retinopathy and may have neovascularization or hemorrhage on retinal exam. Many children will require orthodontia due to maxillary protrusion secondary to compensatory bone marrow expansion (Orthodontics (see NV providers [4])).
Chest
Tachypnea, retractions, decreased breath sounds, crackles, or wheezing should prompt evaluation for acute chest syndrome or reactive airway disease.
Heart
Systolic flow murmurs may be present secondary to chronic anemia. An accentuated pulmonic component of the second heart sound or gallop can indicate development of pulmonary hypertension.
Abdomen
Acute splenomegaly may indicate splenic sequestration crisis. Chronic splenomegaly may result in hypersplenism or put patients at risk for splenic rupture. Due to chronic hemolysis, there is an increased risk for cholelithiasis, which can manifest with a positive Murphy’s sign (hypersensitivity to deep palpation in the subcostal area when a patient with gallbladder disease takes a deep breath) or severe right upper quadrant tenderness.
Genitalia
Extremities/Musculoskeletal
Limited range of motion with forced internal rotation of the lower extremity or painful straight leg raise may be a sign of avascular necrosis of the femoral head. Avascular necrosis in the humeral head may cause similar symptoms in the shoulder. Point tenderness or swelling with fever can occur in the setting of osteomyelitis or bone infarction.
Testing
Sensory Testing
Patients receiving iron chelation should have audiology screening yearly. Testing should also be offered if a patient or parent reports concerns about hearing loss as there is some newer evidence that patients with SCD may have increased risk for hearing deficits. [Schopper: 2019]
Laboratory Testing
Imaging
A one-time baseline MRI/MRA should be obtained for patients with the most severe types of sickle cell disease to evaluate for silent cerebral infarcts and CNS vasculopathy. This screening should occur in early childhood but should be performed when the patient does not require sedation for the imaging study. Baseline screening in adults should also be performed if no previous history of MRI/MRA.
Routine screening with echocardiograms is no longer recommended. Patients with new cardiopulmonary symptoms should receive an echocardiogram and be evaluated for asthma or other non-sickle-cell-disease conditions.
Patients receiving chronic transfusions should be screened annually with MRI for hepatic or cardiac siderosis to determine need for oral chelation therapy.
Genetic Testing
Specialty Collaborations & Other Services
Pediatric Ophthalmology (see NV providers [6])
Pediatric Pulmonology (see NV providers [4])
Pediatric Cardiology (see NV providers [4])
Pediatric Neurology (see NV providers [5])
Genetic Testing and Counseling (see NV providers [11])
Treatment & Management
Overview
Pearls & Alerts for Treatment & Management
HydroxyureaHydroxyurea is the mainstay of therapy for patients with Hb SS disease or sickle beta zero thalassemia. It is recommended that this therapy be offered to these patients starting at 9 months of age. Hydroxyurea can increase fetal hemoglobin levels and has been shown to decrease the frequency of vaso-occlusive complications, increase total hemoglobin levels and decrease number of transfusions needed. Hydroxyurea can cause myelosuppression and the dose should be titrated to prevent excessive neutropenia or thrombocytopenia. [Yawn: 2014]
L-glutamine for sickle cell disease treatmentThe FDA approved the use of L-glutamine powder (Endari) in July 2017 for patients age 5 years and older. It is an oral medication shown to decrease rates of acute chest syndrome, as well as frequency of hospitalization and length of stay for vaso-occlusive pain crises in patients who had 2 or more episodes of pain in the preceding year. It is the first drug to gain FDA-approval for sickle cell disease treatment in more than 2 decades.
Perioperative surgical managementUndergoing general anesthesia for surgical procedures increases the risk for development of acute chest syndrome or vaso-occlusive pain crisis. Preoperative simple or exchange transfusion has been demonstrated to minimize these risks.
Newer therapiesRecent advances in therapy have resulted in dramatic reductions in symptoms and complications of sickle cell disease. Hydroxyurea was approved by the FDA for use in children in 2017; voxeletor and crizanlizumab were both approved in 2019. Hematopoietic stem cell transplantation can be curative. [Gardner: 2018] Gene therapy is currently experimental and clinical trials are ongoing to determine long term safety and efficacy. See under the Hematology/Oncology System below for more detail.
How should common problems be managed differently in children with Sickle Cell Disease?
Viral Infections
Bacterial Infections
Systems
Hematology/Oncology
Children with sickle cell disease should be started on penicillin prophylaxis at diagnosis to prevent pneumococcal sepsis. Prophylaxis may be discontinued at age 5 if there is no history of pneumococcal bacteremia or surgical splenectomy. [Falletta: 1995] All patients should complete a normal immunization series as well as additional meningococcal and pneumococcal vaccines as recommended by the most current version of the Centers for Disease Control and Prevention's Immunization Schedules (CDC).
The 2014 National Heart, Lung, and Blood Institute recommend that hydroxyurea therapy be offered to all patients with homozygous sickle cell anemia or sickle β0 thalassemia starting at 9 months of age regardless of prior complications or baseline fetal hemoglobin percentage. [Yawn: 2014] Hydroxyurea therapy has been shown to decrease frequency of vaso-occlusive pain crises, acute chest syndrome, and lifetime transfusion requirements, as well as increase life expectancy. [Steinberg: 2010] Hydroxyurea for Sickle Cell (Hospital for Sick Children) (
 395 KB) is an
information sheet for parents with children prescribed hydroxyurea
(Hydrea).
395 KB) is an
information sheet for parents with children prescribed hydroxyurea
(Hydrea).Hydroxyurea can cause depression of the absolute neutrophil count. Patients treated with hydroxyurea should be monitored every 3 months with CBCs. The dose is titrated to achieve an absolute neutrophil count between 1,250 and 4,000, with a maximum dose of 35 mg/kg/day. Recommendations for initiation and monitoring of hydroxyurea therapy are summarized in [Yawn: 2014].
Patients with abnormal cerebral velocities measured by transcutaneous Doppler should be treated with chronic red cell transfusions, which has been shown to decrease stroke risk by 90%. [Adams: 1998] New data suggest that children with abnormal TCDs who have had more than 1 year of chronic transfusions AND who have been evaluated with MRI and MRA to assess for cerebral vasculopathy may be safely switched to a maximal tolerated dose of hydroxyurea for primary stroke prophylaxis. [Ware: 2016] Discussion should always be included with SCD specialist.
Voxelotor was granted FDA approval in November 2019 for use in adult and pediatric patients age 12 and older. Voxelotor increases hemoglobin levels and reduces hemolysis by directly binding and stabilizing oxygenated hemoglobin and reducing polymerization. It is taken by mouth once daily and is generally well tolerated with a side effect profile that may include diarrhea and headache. Findings from the recently published phase III trial (HOPE) demonstrated an increase in hemoglobin by >1 g/dL in a significant proportion of participants receiving the drug. A trend toward decreased pain crises was observed, but statistical significance was not achieved. Voxelotor may be taken concurrently with hydroxyurea. There is no data that voxelotor prevents stroke and should not replace chronic transfusions or hydroxyurea in primary or secondary stroke prophylaxis. [Vichinsky: 2019]
The FDA approved crizanlizumab in November 2019 for use in patients 16 and older to reduce the frequency of acute pain crises. In a recent phase II trial (SUSTAIN), crizanlizumab reduced acute pain events by 45.3% and increased the median time to first and second pain episodes. Patients may take crizanlizumab while also taking hydroxyurea. Crizanlizumab is a humanized monoclonal antibody that binds to P-selectin and likely decreases pain crises by reducing vaso-occlusion and inflammation and improving blood flow by inhibiting cellular adhesion to the endothelium. It is given intravenously every 2 weeks for 2 doses, then monthly thereafter. It is generally well-tolerated. Adverse events may include arthralgia, diarrhea, pruritus, vomiting, and chest pain. A number of clinical trials are currently open to further investigate efficacy and safety of crizanlizumab in both adult and pediatric patients. [Ataga: 2017]
Secondary prevention
Patients with prior stroke should begin a chronic transfusion regimen to prevent stroke recurrence. The goal of chronic transfusions is to maintain sickle hemoglobin percentage <30% and maximum hemoglobin <11-12 g/dL. Chronic transfusions are sometimes prescribed for patients with frequent and severe episodes of vaso-occlusive pain or acute chest syndrome.
Iron chelators are prescribed for patients receiving chronic transfusions who have evidence of iron overload. This therapy should be monitored by a sickle cell specialist familiar with this medication. Patients will need annual ophthalmology and audiology screening while taking iron chelators.
L-glutamine was approved by the FDA in July 2017 for patients age 5 years and older. This is the first drug to gain approval for sickle cell disease in more than 2 decades. It is an oral medication taken twice daily and has been shown to decrease rates of acute chest syndrome, as well as frequency of hospitalization and length of stay for vaso-occlusive pain crises in patients who had 2 or more episodes of pain in the preceding year. It is well-tolerated with a minimal side effect profile that may include constipation, nausea, abdominal pain, or headache.
Management of acute complications
Vaso-occlusive pain crisis: Patients presenting with painful crises should receive therapy with NSAIDs and opiates. Moderate to severe episodes may require treatment with parenteral opiates and hospitalization. Intravenous fluid hydration may help decrease red blood cell sickling. The FACES Pain Rating Scale (Wong-Baker Foundation) is a self-assessment that uses expressions on faces to depict pain level. The scale can be used with people ages 3 and older to facilitate communication and improve assessment so pain management can be addressed. Sickle Cell: What to Do When Your Child is Experiencing Pain (Children's Hospital Colorado) (
304 KB) lists steps for comforting a child with sickle cell who is experiencing
pain and includes medication dosing charts.Acute chest syndrome: Patients with respiratory symptoms and a new infiltrate on chest radiography should be admitted and managed by a sickle cell specialist. Concern for pneumonia requires that individuals should be treated empirically with a third-generation cephalosporin and macrolide antibiotic. For patients with hypoxia, red cell transfusion is always indicated. Simple or red cell exchange transfusion may be required for worsening respiratory distress.
Splenic sequestration: Patients should be admitted to the hospital for serial examinations and CBCs. Red cell transfusion may be required.
Sickle Cell: When to Call The Doctor (Children's Hospital Colorado) (
287 KB) lists possible symptoms in children with sickle cell disease that may
indicate a serious problem and need immediate medical
attention.Curative therapy
Patients with sickle cell disease and severe complications may be cured by allogeneic hematopoietic stem cell transplant. The typical indications for transplant are frequent episodic vaso-occlusive pain, recurrent acute chest syndrome, or prior history of stroke. The best candidates for hematopoietic stem cell transplant have an HLA-identical related donor and are less than 16 years of age.
The absence of a suitable donor is one of the major factors limiting the availability of curative therapy for patients with severe disease. Alternative donor transplant using HLA-identical unrelated donors has been attempted but is associated with increased morbidity and mortality due to development of graft versus host disease in many patients limiting wider adoption of this technique. Clinical trials are currently in progress evaluating the use of haploidentical donors with post-transplant cyclophosphamide for graft versus host disease prophylaxis. [Kassim: 2017]
Newer (potential) curative options
Gene therapy is another strategy that may increase the number of patients eligible for curative therapy. There are currently several competing strategies (e.g., gene addition, gene editing) that either increase the concentrations of non-sickling adult hemoglobin or protective fetal hemoglobin. Currently, gene therapy is only available in the United States in clinical trials. [Olowoyeye: 2020] [Demirci: 2018]
Specialty Collaborations & Other Services
Pediatric Hematology/Oncology (see NV providers [7])
Sickle Cell Disease Centers (see NV providers [4])
Audiology (see NV providers [8])
Pediatric Ophthalmology (see NV providers [6])
Neurology
Specialty Collaborations & Other Services
Pediatric Neurology (see NV providers [5])
Neuropsychiatry/Neuropsychology (see NV providers [3])
General Counseling Services (see NV providers [213])
Renal
Specialty Collaborations & Other Services
Pediatric Nephrology (see NV providers [2])
Recreation & Leisure

- Avoid handling reptiles and amphibians (turtles, lizards, frogs, etc.) due to increased risk of Salmonella infection. See Animals that May Carry Salmonella (CDC) for a more extensive list.
- Participate in sports and other physical activities, but remain hydrated at all times.
- Avoid environmental exposure to severe cold, which may precipitate vasoconstriction and painful crises.
- If residing at sea level, do not travel to an altitude above 7,500 feet due to the risk for painful crises and splenic infarction secondary to hypoxia.
Learning/Education/Schools
Transitions
For a successful transition from pediatric to adult sickle cell care, counsel the patient about the transition prior to transfer; communicate among pediatric and adult providers, and ensure that the first visit to an adult provider is before the final pediatric visit or within 1-3 months of the last pediatric visit. [Sobota: 2017] Potential indicators for successful transition are that the:
- Individual is keeping clinic appointments and remaining adherent to treatment and medications
- Written transfer summary has been sent to the adult provider
- First visit to adult provider is within an appropriate interval after leaving pediatric hematology.
- Overall quality of life is good
Ask the Specialist
How do I interpret the newborn screening report, and what should I do if the results are consistent with possible sickle cell disease?
The newborn screening report will list the types of hemoglobin present in
decreasing order of relative quantity. Because fetal hemoglobin is most common
in the first several weeks of life, it is always listed first in untransfused
babies. Normal infants should have a screen showing “F+A”. Patients with one of
the sickling disorders may have newborn screens showing “F+S”, “F+S+A” (sickle
beta plus thalassemia) or “F+S+C”. Patients with “F+A+S” have sickle cell trait.
If a child’s newborn screen results are consistent with
possible sickle cell disease, the state newborn screening lab will provide
instructions for sending a second newborn screen to confirm the abnormal
hemoglobin pattern. If sickle cell disease is confirmed, the patient should be
referred to pediatric hematology or a comprehensive sickle cell center.
Penicillin prophylaxis should be prescribed by two months of age for patients
with severe phenotypes (homozygous sickle cell anemia or sickle beta0
thalassemia) if there is a delay in subspecialty evaluation. Sickle Cell Anemia has further Information
about initial clinical response to a positive newborn screen.
What additional immunizations to patients with sickle cell disease need?
You should consult the latest version of the CDC Immunization Schedule for
high-risk patients with functional or anatomic splenectomy for details as this
may change annually.
Patients with sickle cell disease
should receive annual influenza immunization. Because of the functional
asplenia, they are at increased risk for infection with encapsulated organisms.
Therefore, they should complete the normal pneumococcal series with PCV13 as
well as a dose of PPSV23 after the age of 2 and at least 8 weeks after the last
PCV13. A single booster of PPSV23 should be administered 5 years after the last
dose of PPSV23. Primary meningococcal vaccination should occur early with two
doses given 8 weeks apart starting at age 2 years. They should also receive
meningococcal B vaccination with a 2-dose series starting at 10 years of
age.
What counseling should I perform for a patient with sickle cell trait?
Sickle cell trait is common with a prevalence of up to 1:14 in African Americans.
It is appropriate for primary care providers to provide counseling to patients
regarding their sickle cell trait status. It is important that patients know
they do not have sickle cell disease. Patients with sickle trait rarely have
health problems. In some extreme conditions, such as severe dehydration or high
altitude, there may be complications such as rhabdomyolysis or splenic
infarction. Sometimes patients with sickle trait may have blood in their urine
and should notify their doctor immediately if this
happens.
The National Collegiate Athletic Association
(NCAA) screens every collegiate student-athlete for sickle cell trait. The
presence of sickle cell trait is not a contraindication to participation, but
additional precautions should be taken. Precautions include paying particular
attention to hydration; avoiding high-caffeine energy drinks, supplements, and
stimulants; graduated activity; and stopping activity in the event of symptoms,
such as muscle pain, abnormal weakness, or breathlessness. See Sickle Cell Trait – Fact Sheet for Student Athletes (NCAA) ( 1.3 MB).
Patients should also be made aware of the
reproductive implications of their trait status. If their partner also has
sickle trait or beta-thalassemia trait, then their offspring have a 25% chance
of having sickle cell disease. We encourage patients to discuss their partner’s
trait status prior to conceiving.
Do patients with sickle cell trait experience any symptoms or complications?
In general, individuals with sickle cell trait are asymptomatic and do not experience sickle-related complications like pain. Some data suggest individuals with sickle trait may be at slightly increased risk for renal complications (hematuria), venous thromboembolism (PE) in adulthood, and rarely experience splenic infarction when exposure to altitude. Testing for sickle cell trait is required by the NCAA for college athletes as there have been rare cases of exercise-related complications in individuals who carry sickle cell trait.[Naik: 2015]
Resources for Clinicians
On the Web
Information about initial clinical response to a positive newborn screen for sickle cell disease; Medical Home Portal.
Sickle Cell Information for Healthcare Providers (CDC)
A broad range of useful information, including clinician and patient education materials, clinical guidelines, data/statistics,
and scientific articles; Centers for Disease Control and Prevention
Sickle Cell Disease (GeneReviews)
Detailed information addressing clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular
pathogenesis; from the University of Washington and the National Library of Medicine.
Helpful Articles
PubMed search for articles published in the last year about sickle cell disease in children
Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro
S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP.
Crizanlizumab for the prevention of pain crises in sickle cell disease.
N Engl J Med.
2017;376(5):429-439.
PubMed abstract / Full Text
Borhade MB, Kondamudi NP.
Sickle Cell Crisis.
StatPearls [Internet] Treasure Island (FL) StatPearls Publishing.
2020.
PubMed abstract / Full Text
Piel FB, Steinberg MH, Rees DC.
Sickle Cell Disease.
N Engl J Med.
2017;376(16):1561-1573.
PubMed abstract
Gardner RV.
Sickle cell disease: advances in treatment.
Ochsner J.
2018;18(4):377-389.
PubMed abstract / Full Text
Wang CJ, Kavanagh PL, Little AA, Holliman JB, Sprinz PG.
Quality-of-care indicators for children with sickle cell disease.
Pediatrics.
2011;128(3):484-93.
PubMed abstract
Clinical Tools
Assessment Tools/Scales
FACES Pain Rating Scale (Wong-Baker Foundation)
Self-assessment that uses expressions on faces to depict pain level.
Patient Education & Instructions
Sickle Cell: What to Do When Your Child is Experiencing Pain (Children's Hospital Colorado) ( 304 KB)
Steps for comforting a child with sickle cell who is experiencing pain; includes medication dosing charts and suggestions
for when to call your doctor.
Hydroxyurea for Sickle Cell Disease (American Society of Sickle Cell Disease) ( 876 KB)
Information for parents with children about side effects and instructions for how to administer hydroxyurea, what to do if
a dose is missed, and safety measures to prevent infections.
Teens with Sickle Cell Disease Moving to Adult Care (St. Jude Children’s Research Hospital) ( 456 KB)
Tips for teens taking responsibility for changing from a pediatric hematologist to a hematologist who treats adults. Tips
include practical suggestions for understanding how sickle cell disease affects your body, getting your full medical record,
and obtaining a medical ID card, durable power of attorney for health care, and health insurance.
Resources for Patients & Families
Platt AF, Eckman J, Hsu L.
Hope & Destiny: The Patient and Parent's Guide to Sickle Cell Disease and Sickle Cell Trait.
4th ed. Hilton Publishing;
2016.
098475668X https://www.amazon.com/Hope-Destiny-Patient-Parents-Disease/dp/0984756...
A 260-page book that offers in-depth information about research, treatment, pain management, and preventing complications.
Information on the Web
For Parents & Families
Information for parents to help them better care for their
child with complex conditions from diagnosis through their child's
transition to adult care; Medical Home Portal.
Sickle Cell Disease (MedlinePlus)
Overview of sickle cell disease plus links to many other relevant sources of information and support for patients and families;
from the National Library of Medicine.
Sickle Cell Disease (CDC)
Comprehensive information about sickle cell disease for caregivers, families, and patients; Centers for Disease Control and
Prevention.
Sickle cell anemia: How our red blood cells evolved to fight malaria (HealthMatch)
This web resource discusses the evolutionary role of how sickle cells help fight malaria, with a discussion of emerging therapeutics
and option to search for relevant drug trials.
National & Local Support
Sickle Cell Disease Association of America
The mission of this nonprofit is to improve the quality of health, life, and services for individuals, families, and communities
affected by sickle cell disease and related conditions while promoting the search for a cure.
Studies/Registries
Sickle Cell Disease (ClinicalTrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Services for Patients & Families in Nevada (NV)
| Service Categories | # of providers* in: | NV | NW | Other states (3) (show) | | NM | RI | UT |
|---|---|---|---|---|---|---|---|---|
| Audiology | 8 | 3 | 22 | 24 | 22 | |||
| General Counseling Services | 213 | 1 | 10 | 30 | 298 | |||
| Genetic Testing and Counseling | 11 | 5 | 5 | 7 | 10 | |||
| Neuropsychiatry/Neuropsychology | 3 | 1 | 9 | 6 | ||||
| Orthodontics | 4 | 3 | 17 | |||||
| Pediatric Cardiology | 4 | 3 | 17 | 4 | ||||
| Pediatric Hematology/Oncology | 7 | 2 | 4 | 11 | 4 | |||
| Pediatric Nephrology | 2 | 2 | 10 | 1 | ||||
| Pediatric Neurology | 5 | 5 | 18 | 8 | ||||
| Pediatric Ophthalmology | 6 | 1 | 6 | 8 | 4 | |||
| Pediatric Pulmonology | 4 | 4 | 6 | 3 | ||||
| Sickle Cell Disease Centers | 4 | 1 | 1 | 1 | 2 | |||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Adams R, McKie V, Nichols F, Carl E, Zhang DL, McKie K, Figueroa R, Litaker M, Thompson W, Hess D.
The use of transcranial ultrasonography to predict stroke in sickle cell disease.
N Engl J Med.
1992;326(9):605-10.
PubMed abstract
Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla
D.
Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler
ultrasonography.
N Engl J Med.
1998;339(1):5-11.
PubMed abstract
Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro
S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP.
Crizanlizumab for the prevention of pain crises in sickle cell disease.
N Engl J Med.
2017;376(5):429-439.
PubMed abstract / Full Text
Blinder MA, Duh MS, Sasane M, Trahey A, Paley C, Vekeman F.
Age-related emergency department reliance in patients with sickle cell disease.
J Emerg Med.
2015;49(4):513-522.e1.
PubMed abstract
Borhade MB, Kondamudi NP.
Sickle Cell Crisis.
StatPearls [Internet] Treasure Island (FL) StatPearls Publishing.
2020.
PubMed abstract / Full Text
Brandow AM, Carroll CP, Creary S, Edwards-Elliott R, Glassberg J, Hurley RW, Kutlar A, Seisa M, Stinson J, Strouse JJ, Yusuf
F, Zempsky W, Lang E.
American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain.
Blood Adv.
2020;4(12):2656-2701.
PubMed abstract / Full Text
Chou S, Alsawas M, Fasano R, Field J, Hendrickson J, Howard J, Kameka M, Kwiatkowski J, Pirenne F, Shi P, Stowell S, Thein
S, Westhoff C, Wong T, Akl E.
American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support.
Blood Adv..
2020;4(2):327-355.
/ Full Text
Coates TD, Wood JC.
How we manage iron overload in sickle cell patients.
Br J Haematol.
2017;177(5):703-716.
PubMed abstract / Full Text
DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, Meier ER, Howard TH, Majumdar S, Inusa BP, Telfer PT,
Kirby-Allen M, McCavit TL, Kamdem A, Airewele G, Woods GM, Berman B, Panepinto JA, Fuh BR, Kwiatkowski JL, King AA, Fixler
JM, Rhodes MM, Thompson AA, Heiny ME, Redding-Lallinger RC, Kirkham FJ, Dixon N, Gonzalez CE, Kalinyak KA, Quinn CT, Strouse
JJ, Miller JP, Lehmann H, Kraut MA, Ball WS Jr, Hirtz D, Casella JF.
Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia.
N Engl J Med.
2014;371(8):699-710.
PubMed abstract / Full Text
DeBaun MR, Jordan LC, King AA, Schatz J, Vichinsky E, Fox CK, McKinstry RC, Telfer P, Kraut MA, Daraz L, Kirkham FJ, Murad
MH.
American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular
disease in children and adults.
Blood Adv.
2020;4(8):1554-1588.
PubMed abstract / Full Text
Demirci S, Uchida N, Tisdale JF.
Gene therapy for sickle cell disease: An update.
Cytotherapy.
2018;20(7):899-910.
PubMed abstract / Full Text
Falletta JM, Woods GM, Verter JI, Buchanan GR, Pegelow CH, Iyer RV, Miller ST, Holbrook CT, Kinney TR, Vichinsky E.
Discontinuing penicillin prophylaxis in children with sickle cell anemia. Prophylactic Penicillin Study II.
J Pediatr.
1995;127(5):685-90.
PubMed abstract
Gardner RV.
Sickle cell disease: advances in treatment.
Ochsner J.
2018;18(4):377-389.
PubMed abstract / Full Text
Hassell KL.
Population estimates of sickle cell disease in the U.S.
Am J Prev Med.
2010;38(4 Suppl):S512-21.
PubMed abstract
Hemker BG, Brousseau DC, Yan K, Hoffmann RG, Panepinto JA.
When children with sickle-cell disease become adults: lack of outpatient care leads to increased use of the emergency department.
Am J Hematol.
2011;86(10):863-5.
PubMed abstract
Kassim AA, Sharma D.
Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape.
Hematol Oncol Stem Cell Ther.
2017;10(4):259-266.
PubMed abstract
Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky
EP.
Sickle cell disease.
Nat Rev Dis Primers.
2018;4:18010.
PubMed abstract
Liem RI, Lanzkron S, D Coates T, DeCastro L, Desai AA, Ataga KI, Cohen RT, Haynes J, Osunkwo I, Lebensburger JD, Lash JP,
Wun T, Verhovsek M, Ontala E, Blaylark R, Alahdab F, Katabi A, Mustafa RA.
American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease.
Blood Adv.
2019;3(23):3867-3897.
PubMed abstract / Full Text
Lorey FW, Arnopp J, Cunningham GC.
Distribution of hemoglobinopathy variants by ethnicity in a multiethnic state.
Genet Epidemiol.
1996;13(5):501-12.
PubMed abstract
Minniti CP, Eckman J, Sebastiani P, Steinberg MH, Ballas SK.
Leg ulcers in sickle cell disease.
Am J Hematol.
2010;85(10):831-3.
PubMed abstract / Full Text
Naik RP, Haywood C Jr.
Sickle cell trait diagnosis: clinical and social implications.
Hematology Am Soc Hematol Educ Program.
2015;2015:160-7.
PubMed abstract / Full Text
Olowoyeye A, Okwundu CI.
Gene therapy for sickle cell disease.
Cochrane Database Syst Rev.
2020;11:CD007652.
PubMed abstract
Piel FB, Steinberg MH, Rees DC.
Sickle Cell Disease.
N Engl J Med.
2017;376(16):1561-1573.
PubMed abstract
Platt AF, Eckman J, Hsu L.
Hope & Destiny: The Patient and Parent's Guide to Sickle Cell Disease and Sickle Cell Trait.
4th ed. Hilton Publishing;
2016.
098475668X https://www.amazon.com/Hope-Destiny-Patient-Parents-Disease/dp/0984756...
A 260-page book that offers in-depth information about research, treatment, pain management, and preventing complications.
Rosen CL, Debaun MR, Strunk RC, Redline S, Seicean S, Craven DI, Gavlak JC, Wilkey O, Inusa B, Roberts I, Goodpaster RL, Malow
B, Rodeghier M, Kirkham FJ.
Obstructive sleep apnea and sickle cell anemia.
Pediatrics.
2014;134(2):273-81.
PubMed abstract / Full Text
Schatz J, Brown RT, Pascual JM, Hsu L, DeBaun MR.
Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease.
Neurology.
2001;56(8):1109-11.
PubMed abstract
Schopper HK, D'Esposito CF, Muus JS, Kanter J, Meyer TA.
Childhood Hearing Loss in Patients With Sickle Cell Disease in the United States.
J Pediatr Hematol Oncol.
2019;41(2):124-128.
PubMed abstract
Sobota AE, Shah N, Mack JW.
Development of quality indicators for transition from pediatric to adult care in sickle cell disease: A modified Delphi survey
of adult providers.
Pediatr Blood Cancer.
2017;64(6).
PubMed abstract
Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, Swerdlow P, Kutlar A, DeCastro L, Waclawiw
MA.
The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up.
Am J Hematol.
2010;85(6):403-8.
PubMed abstract / Full Text
Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, Hassab H, Achebe MM, Alkindi S, Brown RC, Diuguid DL, Telfer
P, Tsitsikas DA, Elghandour A, Gordeuk VR, Kanter J, Abboud MR, Lehrer-Graiwer J, Tonda M, Intondi A, Tong B, Howard J.
A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease.
N Engl J Med.
2019;381(6):509-519.
PubMed abstract
Wang CJ, Kavanagh PL, Little AA, Holliman JB, Sprinz PG.
Quality-of-care indicators for children with sickle cell disease.
Pediatrics.
2011;128(3):484-93.
PubMed abstract
Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, Odame I, Fuh B, George A, Owen W, Luchtman-Jones L, Rogers ZR,
Hilliard L, Gauger C, Piccone C, Lee MT, Kwiatkowski JL, Jackson S, Miller ST, Roberts C, Heeney MM, Kalfa TA, Nelson S, Imran
H, Nottage K, Alvarez O, Rhodes M, Thompson AA, Rothman JA, Helton KJ, Roberts D, Coleman J, Bonner MJ, Kutlar A, Patel N,
Wood J, Piller L, Wei P, Luden J, Mortier NA, Stuber SE, Luban NL, Cohen AR, Pressel S, Adams RJ.
Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle
cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial.
Lancet.
2016;387(10019):661-70.
PubMed abstract
Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe
PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J.
Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members.
JAMA.
2014;312(10):1033-48.
PubMed abstract
Subscription required for full text.
Zemel BS, Kawchak DA, Ohene-Frempong K, Schall JI, Stallings VA.
Effects of delayed pubertal development, nutritional status, and disease severity on longitudinal patterns of growth failure
in children with sickle cell disease.
Pediatr Res.
2007;61(5 Pt 1):607-13.
PubMed abstract